Pediatrics

Inflammation of the uvula is known as uvulitis. Your uvula will appear red, puffy, and larger than normal. Other symptoms of uvulitis may include: itching burning a sore throat spots on your throat snoring difficulty swallowing trouble breathing If you have a swollen uvula along with a fever or abdominal pain, consult with your doctor right away. In rare cases, the uvula can swell enough to block your airway. Swelling of the throat is a life-threatening event. If this happens, seek immediate medical attention. What causes a swollen uvula? Causes Inflammation is your body’s response when it’s under attack. Triggers for inflammation include: environmental and lifestyle factors an infection trauma genetics Environmental and Lifestyle Factors The most common food allergies are peanuts tree nuts milk eggs wheat soy fish, including shellfish You could be having an allergic reaction to something you touched, swallowed, or breathed in. Some common allergens include: food irritants , such as dust, animal dander, or pollen medication exposure to chemicals or other toxic substances, including tobacco Infection You can get viral infections or bacterial infections. Examples of viral infections include: the common cold the flu mononucleosis chickenpox measles croup The most common bacterial infection is strep throat, which occurs due to Streptococcus pyogenes, which is a type of group A Streptococcus. If you have infected tonsils, or tonsillitis, severe inflammation can cause them to push against and irritate your uvula. Trauma Trauma to the uvula can happen if you need an intubation, such as during surgery. Your uvula can also be injured during a tonsillectomy. This is a procedure to remove your tonsils, which are located on both sides of your uvula. Your throat and uvula can also become irritated if you have acid reflux disease or if you vomit frequently. Genetics A condition called hereditary angioedema (HAE) can cause swelling of the uvula and throat, as well as swelling of the face, hands, and feet. Other symptoms include nausea, vomiting, and abdominal pain. It’s an uncommon genetic mutation that occurs in 1 in 10,000 to 1 in 50,000 people. It’s rare, but there are case reports of individuals who have an elongated uvula, which can also interfere with breathing. What are the risk factors for a swollen uvula? Risk Factors Anyone can get uvulitis, but adults get it less often than children do. You’re at increased risk if you: have allergies use tobacco products are exposed to chemicals and other irritants in the environment have a weakened immune system, making you more susceptible to infections How is a swollen uvula diagnosed? Diagnosis If you have fever or swelling of your throat, see your doctor. Be prepared to give a complete medical history. Tell your doctor: about all the over-the-counter and prescription medications you take if you’re a smoker or you chew tobacco if you’ve recently tried new foods if you’ve been exposed to chemicals or unusual substances about your other symptoms, such as abdominal pain, fever, or dehydration Your doctor may be able to make a diagnosis through a physical exam. It’s likely you’ll also need a throat swab to evaluate for strep or to obtain secretions for culture to determine if you have another bacterial or fungal infection. This test is known as the rapid strep test. You may also need a nasal swab to test for influenza. Blood testing can help identify or rule out some other infectious agents. If those tests are inconclusive, you may need to see an allergist. Blood and skin tests can help identify foods or other substances that cause a reaction. Learn more: Allergy testing » If necessary, imaging tests can provide a more detailed view of your throat and the surrounding area. What’s the treatment for a swollen uvula? Treatment When you have something like the common cold, swelling usually clears up on its own without treatment. Otherwise, treatment will depend on how severe your symptoms are, as well as what’s causing the inflammation. Infection Viral infections tend to clear up without treatment. The only upper respiratory infection for which an antiviral medication is available is influenza. Antibiotics can treat bacterial infections. Even after symptoms clear up, take all the medication as prescribed. If your condition may be contagious, stay home until your doctor tells you that you’re no longer at risk of spreading it to others. Allergy If you test positive for an allergy, try to avoid the allergen in the future. Doctors usually treat allergies with antihistamines or steroids. Anaphylaxis is a severe allergic reaction. Doctors use epinephrine to treat this reaction. Hereditary angioedema Your doctor may treat HAE with any of the following: anabolic steroids, or androgens antifibrinolytics C1 inhibitors, such as C1 esterase inhibitor (Berinert) or C1 esterase inhibitor (recombinant) (Ruconest) a plasma kallikrein inhibitor, such as ecallantide (Kalbitor) bradykinin receptor antagonist, such as icatibant injection (Firazyr) Tell your doctor if you have new or worsening symptoms, and follow up as necessary. Tips for relief home treatment If you have a swollen uvula or sore throat, it’s your body’s way of telling you that something is wrong. A few home remedies can help keep you strong and soothe your irritated throat. Make sure you’re getting enough fluids. If your throat hurts when you drink, try drinking small amounts throughout the day. Your urine should be light in color. If it’s dark yellow or brown, you’re not drinking enough and may be dehydrated. Additional tips include the following: Cool your throat by sucking on ice chips. Frozen juice bars or ice cream may also do the trick. Gargle with warm salt water to ease your dry, scratchy throat. Aim for a full night’s sleep, and nap during the day if you can. What’s the outlook? Outlook A swollen uvula isn’t a common occurrence. Most of the time it clears up without treatment. If you have an infection, prompt treatment should take care of the problem within a week or two. If you have allergies that lead to swelling of the uvula or throat, do your best to avoid that allergen. You should also be prepared to deal with an attack if you come into contact with the substance again. If you’ve ever had anaphylaxis, ask your doctor if you should carry injectable epinephrine (EpiPen) in case of emergency. People with HAE must learn to recognize triggers and early warning signs of an attack. Talk to your doctor about how to manage HAE. Article Resources Was this article helpful?Yes No Share Tweet Email Print Read This Next 9-Month-Old Baby: Developmental Milestones and Guidelines 9-Month-Old Baby: Developmental Milestones and Guidelines Read More » All of the ‘Firsts’ That Come with Breast-Feeding All of the ‘Firsts’ That Come with Breast-Feeding Read More » 5 Types of Health Professionals You Should Know About 5 Types of Health Professionals You Should Know About Read More » What’s the Difference Between a Fracture and a Break? What’s the Difference Between a Fracture and a Break? Read More » Is Corn a Vegetable? Is Corn a Vegetable? Read More » Advertisement Advertisement Advertisement

Strep throat is a bacterial infection that can make your throat feel sore and scratchy. Strep throat accounts for only a small portion of sore throats. If untreated, strep throat can cause complications, such as kidney inflammation or rheumatic fever. Rheumatic fever can lead to painful and inflamed joints, a specific type of rash or heart valve damage. Strep throat is most common in children, but it affects people of all ages. If you or your child has signs or symptoms of strep throat, see your doctor for prompt testing and treatment.

Shingles is a viral infection that causes a painful rash. Although shingles can occur anywhere on your body, it most often appears as a single stripe of blisters that wraps around either the left or the right side of your torso. Shingles is caused by the varicella-zoster virus — the same virus that causes chickenpox. After you've had chickenpox, the virus lies inactive in nerve tissue near your spinal cord and brain. Years later, the virus may reactivate as shingles. While it isn't a life-threatening condition, shingles can be very painful. Vaccines can help reduce the risk of shingles, while early treatment can help shorten a shingles infection and lessen the chance of complications.
The Zika virus, first identified in Uganda in 1947, is transmitted by the same type of mosquito that carries dengue fever, yellow fever, and chikungunya virus. A mosquito bites an infected person and then passes those viruses to other people it bites. Outbreaks did not occur outside of Africa until 2007, when it spread to the South Pacific.
Cystic fibrosis (CF) is a multisystem disease affecting the digestive system, sweat glands, upper and lower respiratory tracts, and the reproductive tract, but progressive lung disease continues to be the major cause of morbidity and mortality [1]. CF is characterized by abnormal transport of chloride and sodium across the respiratory epithelium, resulting in thickened, viscous airway secretions [2]. Over a highly variable time course ranging from months to decades after birth, individuals eventually develop chronic infection of the respiratory tract with a characteristic array of bacterial flora [3], leading to progressive respiratory insufficiency and eventual respiratory failure. The rate of progression varies widely, depending in part on genotype (including gene modifiers) as well as environmental factors. Registry data from CF Centers in the United States, Canada, and Europe indicate a median survival of about 41 years [4]. Females with CF appear to have higher morbidity and mortality than males [5]. This "gender gap" is modest but consistent across many populations and is hypothesized to be due to the pro-inflammatory effects of estrogens.
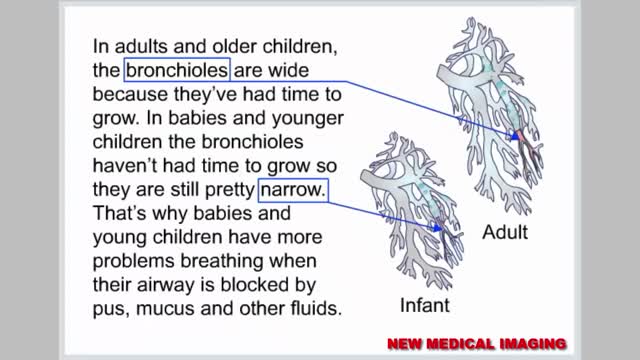
Respiratory syncytial virus (RSV) is a virus that causes infections of the lungs and respiratory tract. It's so common that most children have been infected with the virus by age 2. Respiratory syncytial (sin-SISH-ul) virus can also infect adults. In adults and older, healthy children, the symptoms of respiratory syncytial virus are mild and typically mimic the common cold. Self-care measures are usually all that's needed to relieve any discomfort. Infection with respiratory syncytial virus can be severe in some cases, especially in premature babies and infants with underlying health conditions. RSV can also become serious in older adults, adults with heart and lung diseases, or anyone with a very weak immune system (immunocompromised).
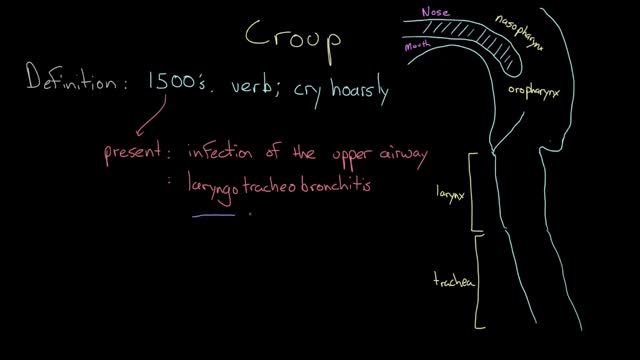
Croup refers to an infection of the upper airway, which obstructs breathing and causes a characteristic barking cough. The cough and other symptoms of croup are the result of swelling around the vocal cords (larynx), windpipe (trachea) and bronchial tubes (bronchi). When a cough forces air through this narrowed passage, the swollen vocal cords produce a noise similar to a seal barking. Likewise, taking a breath often produces a high-pitched whistling sound (stridor). Croup typically occurs in younger children. Croup usually isn't serious and most cases can be treated at home.

Asplenia is the absence of spleen and/or its functions. Abnormalities of the spleen may be classified on a pattern oriented approach, based on splenic imaging.[1] These include anomalies of the following: Shape (clefts, notches, lobules) Location (wandering spleen) Number (asplenia, polysplenia) Size (splenomegaly, atrophy) Solitary lesions (cysts, lymphangiomas, hemangiomas, hamartomas) Multiple lesions (trauma, infections, neoplasms, storage disorders) Diffuse disease (infarction, heavy metal deposition, peliosis) Absence of splenic tissue can be total (congenital asplenia) or partial (hypoplastic) from birth. Loss of splenic tissue due to surgical removal may occur later in life as a result of trauma that causes rupture of the organ. The spleen may be removed in other conditions (eg, hemoglobinopathies) to improve the red cell life expectancy. Removal of the spleen may be undertaken as a result of being involved in a neoplastic processor as a staging procedure in some cancers. Occasionally, the spleen may be removed to address the sheer mass effect of a massive enlargement (such as in storage disorders), which can cause mass effects. Autosplenectomy is the process where the spleen loses its function due to multiple and repeated infarctive episodes, as in sickle hemoglobinopathies. See the image below.
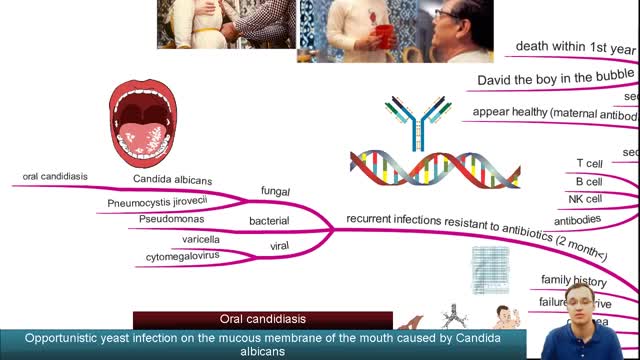
Severe combined immunodeficiency (SCID) is a life-threatening syndrome of recurrent infections, diarrhea, dermatitis, and failure to thrive. It is the prototype of the primary immunodeficiency diseases and is caused by numerous molecular defects that lead to severe compromise in the number and function of T cells, B cells, and occasionally natural killer (NK) cells. Clinically, most patients present before age 3 months. Without intervention, SCID usually results in severe infection and death in children by age 2 years. A committee of experts, initially sponsored by the World Health Organization (WHO), meets every 2 years with the goal to classify the group of primary immunodeficiency diseases according to current understanding of the pathways that become defective in the immune system.[1] Eight classification groups have been determined, with SCID being one of the best studied. Over the past few decades, the diverse molecular genetic causes of SCID have been identified with progress from studies of the immune system.[2] SCID is considered a pediatric emergency because survival depends on expeditious stem cell reconstitution, usually by bone marrow transplantation (BMT). Appropriate diagnosis is essential because instituting proper treatment is lifesaving. Despite the heterogeneity in the pathogenesis of immune defects, common cutaneous manifestations and typical infections can provide clinical clues in diagnosing this pediatric emergency.[3] Skin manifestations were prevalent in primary immunodeficiency disorders studied in 128 pediatric patients in Kuwait; skin infections were the most prevalent findings, seen in 39 patients (30%), followed by dermatitis in 24 (19%).[4] Skin infections were significantly more prevalent in those with congenital defects in phagocyte number, function, or both, as well as in those with well-defined immunodeficiencies. Dermatitis was evident in all patients with hyper–immunoglobulin (Ig) E syndrome and Wiskott-Aldrich syndrome.[4] Erythroderma of infancy with diffuse alopecia was seen exclusively in patients with SCID disorders, and telangiectasia in patients with ataxia telangiectasia; and partial albinism with silvery gray hair was associated with Chediak-Higashi syndrome. With the advances in BMT and gene therapy, patients now have a better likelihood of developing a functional immune system in a previously lethal genetic disease. However, once an infant develops serious infections, intervention is rarely successful.
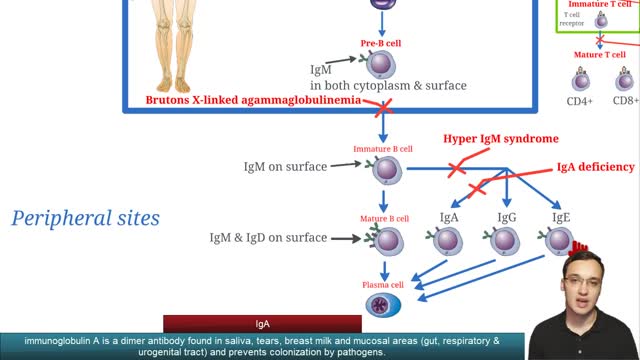
Selective immunoglobulin A deficiency (SIgAD) is a primary immunodeficiency disease and is the most common of the primary antibody deficiencies.[1] Total immunoglobulin A deficiency (IgAD) is defined as an undetectable serum immunoglobulin A (IgA) level at a value < 5 mg/dL (0.05 g/L) in humans. Partial IgAD refers to detectable but decreased IgA levels that are more than 2 standard deviations below normal age-adjusted means.[2, 3] IgAD is commonly associated with normal B lymphocytes in peripheral blood, normal CD4+ and CD8+ T cells, and, usually, normal neutrophil and lymphocyte counts. Anti-IgA autoantibodies of the IgG and/or IgE isotype may be present. Peripheral blood may also be affected by autoimmune cytopenias, eg, autoimmune thrombocytopenia,[4, 5] and patients may have other autoimmune phenomena. IgA was first identified by Graber and Williams in 1952; ten years later, the first patients with IgAD were described. IgAD is a heterogeneous disorder, and the results of intensive study are beginning to elucidate genetic loci and molecular pathogenesis that contribute to various subtypes of this disorder. Several lines of evidence suggest that, in many cases, IgAD and common variable immunodeficiency (CVID) have a common pathogenesis, which is discussed further in Pathophysiology. Other data indicate different genetic risk factors. Family studies show variable inheritance patterns. Familial inheritance of IgAD occurs in approximately 20% of cases,[6] and, within families, IgAD and CVID are associated.[7, 8] Many IgAD patients are asymptomatic (ie, "normal" blood donors) and are identified by finding a laboratory abnormality, without any apparent associated clinical disease. Some patients with IgAD may have the following associated conditions: (1) deficits in one or more immunoglobulin G (IgG) subclasses (this accounts for 20-30% of IgA-deficient patients, many of whom may have total IgG levels within the normal range) or (2) a deficient antibody response to pneumococcal immunization (specific polysaccharide antibody deficiency [SPAD]). Some patients with IgAD later develop CVID, and family members of patients with CVID may have only selective IgAD. Characterization of the receptor for the transmembrane activator and calcium-modulator and cyclophilin ligand interactor (TACI), encoded by the gene TNFRSF13B ( tumor necrosis factor receptor superfamily member 13B), suggests that people with the C104, A181E, and ins204A variants may be at risk for IgAD that progresses to CVID.[9] Primary IgAD is permanent, and below-normal levels have been noted to remain static and persist after 20 years of observation.[10] A recent report documents a rare case of reversion.[11] Environmental factors such as drugs or infections can cause IgAD, but this form is reversible in more than half the cases (see Causes). Although individuals with IgAD have largely been considered healthy, recent studies indicate a higher rate of symptoms. A 20-year follow-up study that compared 204 healthy blood donors with incidentally identified IgAD to 237 healthy subjects with normal IgA levels demonstrated that 80% of IgAD donors and 50% of control subjects had episodes of infections, drug allergy, or autoimmune or atopic disease. Severe respiratory tract infections occurred in 26% of IgAD subjects, in 24% of subjects with decreased IgA levels, and in 8% of control subjects; however, the incidence of life-threatening infections was not increased. IgAD is more common in adult patients with chronic lung disease than in healthy age-matched control subjects.[12] Patients with IgAD are at some increased risk of developing severe reactions after receiving blood products.[13, 14, 15] IgG anti-IgA antibodies may cause severe transfusion reactions if patients with IgAD are given whole blood; therefore, IgA-poor blood or washed red cells are preferred for those patients. IgA-deficient patients with immunoglobulin E (IgE)–class anti-IgA antibodies are at risk for anaphylaxis if they receive blood or intravenous immunoglobulin, but this situation is extremely rare. Individuals with such an unusual profile should receive only low IgA intravenous immunoglobulin preparations. However, caution must be used when administering IGIV to patients with IgAD if their anti-IgA status is unknown. A history devoid of previous blood product administration does not exclude the possibility of anti-IgA antibodies or adverse reactions. Fortunately, appropriate precautions can significantly reduce morbidity (see Treatment). Blood banks can use a simple ELISA screening approach to establish an IgAD blood donor poo
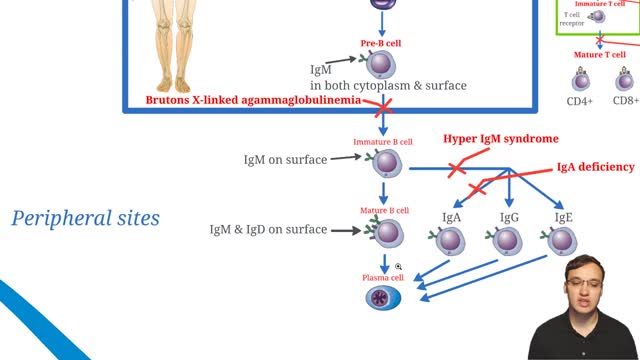
X-linked agammaglobulinemia (XLA), or Bruton agammaglobulinemia, is an inherited immunodeficiency disease caused by mutations in the gene coding for Bruton tyrosine kinase (BTK). The disease was first elucidated by Bruton in 1952, for whom the gene is named. BTK is critical to the maturation of pre–B cells to differentiating mature B cells. The BTK gene defect has been mapped to the long arm of the X chromosome at band Xq21.3 to Xq22, spanning 37.5kb with 19 exons forming 659 amino acids to complete the BTK cytosolic tyrosine kinase. A database of BTK mutations (BTKbase: Mutation registry for X-linked agammaglobulinemia) lists 544 mutation entries from 471 unrelated families showing 341 unique molecular events. No single mutation accounts for more than 3% of mutations in patients. In addition to mutations, a number of variants or polymorphisms have been found.
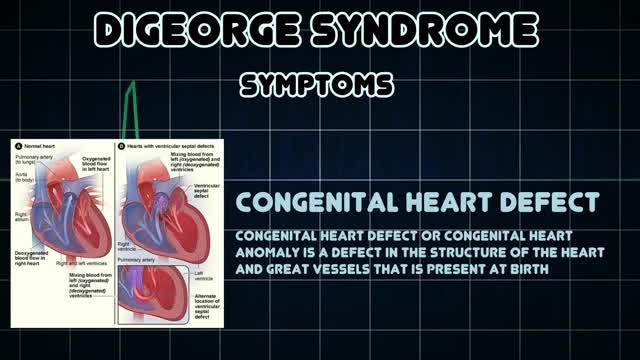
DiGeorge syndrome, also called 22q11.2 deletion syndrome, is a disorder caused by a defect in chromosome 22. It results in the poor development of several body systems. Medical problems commonly associated with DiGeorge syndrome include heart defects, poor immune system function, a cleft palate, complications related to low levels of calcium in the blood, and delayed development with behavioral and emotional problems. The number and severity of symptoms associated with DiGeorge syndrome vary greatly. However, almost everyone with DiGeorge syndrome needs treatment from specialists in a variety of fields. Before the discovery of the chromosome 22 defect, the disorder was known by several names — DiGeorge syndrome, velocardiofacial syndrome, Shprintzen syndrome, CATCH22 and others. Although the term "22q11.2 deletion syndrome" is frequently used today — and is generally a more accurate description — previous names for the disorder are still used.

What causes rheumatic fever? Rheumatic fever is not an infection itself, but rather the result of an untreated strep infection. When your body senses the strep infection, it sends antibodies to fight it. Sometimes, these antibodies attack the tissues of your joints or heart instead. If the antibodies attack your heart, they can cause your heart valves to swell, which can lead to scarring of the valve "doors" (called leaflets or cusps). Who is at risk for rheumatic fever? Fewer than 0.3% of people who have strep throat also get rheumatic fever. Rheumatic fever is most common among children aged 5 to 15, but adults may have the condition as well. Doctors think that a weakened immune system may make some people more likely to get rheumatic fever. And, although antibiotic medicines have reduced the number of cases of rheumatic fever in developed countries, there are still thousands of reported cases. What are the symptoms of rheumatic fever and how is it diagnosed? Symptoms of rheumatic fever usually begin 1 to 6 weeks after you have had a strep infection. They are Fever Joint pain or swelling in your wrists, elbows, knees, or ankles Small bumps under the skin over your elbows or knees (called nodules) A raised, red rash on your chest, back, or stomach Stomach pain or feeling less hungry Weakness, shortness of breath, or feeling very tired Your doctor will begin by doing a throat culture to find out if you have a strep infection. Then, your doctor will use a stethoscope to listen to your heart. He or she will also look for nodules on your joints. Sometimes, blood tests, chest x-rays, or an electrocardiogram (ECG or EKG) may be needed for a more definite diagnosis. How is rheumatic fever treated? Rheumatic fever must be treated right away. If you have a sore throat that lasts longer than 3 days, or if you have a fever and headache along with your sore throat, you should see your doctor for a throat culture. Even if you do not have a sore throat but have a fever and a skin rash, this could also mean a strep infection, and you should get tested. Remember rheumatic fever can result from an untreated strep infection, so it is very important to treat the infection before it leads to a worse condition.
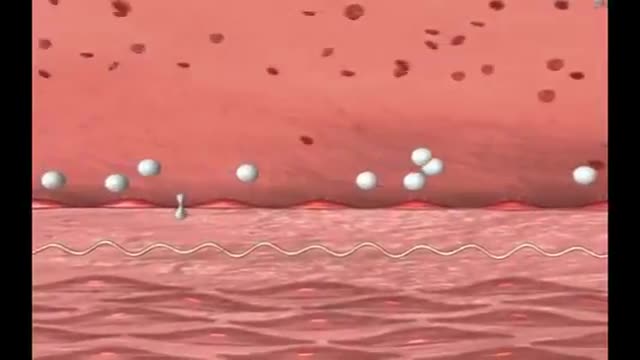
Kawasaki disease is a condition that causes inflammation in the walls of medium-sized arteries throughout the body, including the coronary arteries, which supply blood to the heart muscle. Kawasaki disease is also called mucocutaneous lymph node syndrome because it also affects lymph nodes, skin, and the mucous membranes inside the mouth, nose and throat. Signs of Kawasaki disease, such as a high fever and peeling skin, can be frightening. The good news is that Kawasaki disease is usually treatable, and most children recover from Kawasaki disease without serious problems.

Twin girls joined at the head who share the same brain and so much more

Toilet Training Boys, Training Potty, Best Way To Potty Train, What Age Do You Potty Train
http://potty-training-fast.good-info.co
Wanna have some fun imagining life without diapers?
Imagine if your child would disappear on their own
one minute and all of a sudden the next minute you
hear the toilet flush and the sink start to run.
Can you imagine it?
Life becomes so much easier the second your child
becomes potty trained and you start to wonder why
you didn't just get it over with sooner...
Would you start potty training right this weekend
if I handed you a guide that guaranteed to get your
child out of diapers in just 3 days?
Click the link below to check it out
http://potty-training-fast.good-info.co
Subscribe to our channel
http://potty-training-fast.blogspot.com/
https://www.youtube.com/watch?v=ck-4RTvP5F4
Toilet Training Boys, Training Potty, Best Way To Potty Train, What Age Do You Potty Train,
3day potty training,
toilet training tips for girls,
how to do potty training,
best potty training book,
potty training boys the easy way,
potty training activities,
how to potty training,
potty training video for toddlers,
when do you potty train,
how to potty train a kid,
potty training at 18 months,
havanese potty training,
how to potty train your baby,
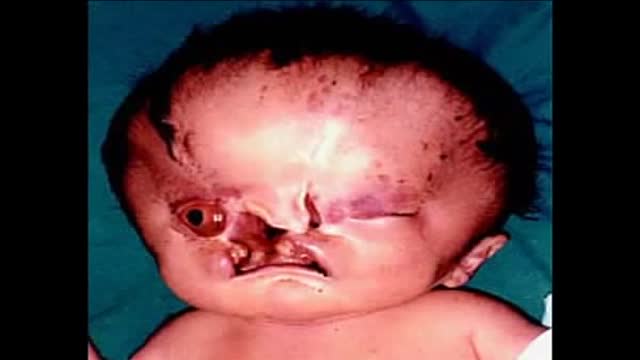
Holoprosencephaly (HPE, once known as arhinencephaly) is a cephalic disorder in which the prosencephalon (the forebrain of the embryo) fails to develop into two hemispheres. Normally, the forebrain is formed and the face begins to develop in the fifth and sixth weeks of human pregnancy. The condition also occurs in other species, as with Cy, the Cyclops kitten.

Gowers' sign is a medical sign that indicates weakness of the proximal muscles, namely those of the lower limb. The sign describes a patient that has to use his hands and arms to "walk" up his own body from a squatting position due to lack of hip and thigh muscle strength. It is named for William Richard Gowers. Gowers' sign is classically seen in Duchenne muscular dystrophy, but also presents itself in centronuclear myopathy, myotonic dystrophy and various other conditions associated with proximal muscle weakness. For this maneuver, the patient is placed on the floor away from any objects that could otherwise be used to pull oneself to a standing position. It is also used in testing paraplegia.

An InterActive Medical Technologies Training Video
QuikRead CRP is a quantitative assay of CRP (C-reactive protein) in whole blood, serum or plasma, using the QuikRead® 101 Instrument and is FDA cleared.
Measurement of CRP helps to evaluate the acute inflammatory processes induced by infectious microbial agents or non-infectious inflammatory stimuli. For in vitro diagnostic use.
QuikRead CRP is not intended for measurement of CRP as a risk marker for coronary heart disease.
For more information visit is at interactivemedtech.net

Multiple strains put your children and teens at risk of meningococcal meningitis. How-to ensure they are fully protected.
