- Physical Examination
- Surgical Examination
- Ophthalmology
- Clinical Skills
- Orthopedics
- Surgery Videos
- Laparoscopy
- Pediatrics
- Funny Videos
- Cardiothoracic Surgery
- Nursing Videos
- Plastic Surgery
- Otorhinolaryngology
- Histology and Histopathology
- Neurosurgery
- Dermatology
- Pediatric Surgery
- Urology
- Dentistry
- Oncology and Cancers
- Anatomy Videos
- Health and Fitness
- Radiology
- Anaesthesia
- Physical Therapy
- Pharmacology
- Interventional Radiology
- Cardiology
- Endocrinology
- Gynecology
- Emergency Medicine
- Psychiatry and Psychology
- Childbirth Videos
- General Medical Videos
- Nephrology
- Physiology
- Diet and Food Health
- Diabetes Mellitus
- Neurology
- Women Health
- Osteoporosis
- Gastroenterology
- Pulmonology
- Hematology
- Rheumatology
- Toxicology
- Nuclear Medicine
- Infectious Diseases
- Vascular Disease
- Reproductive Health
- Burns and Wound Healing
- Other
Latest videos

French Insuficiencia cardiaca

Femoral Venous Line Placement

Austin lip augmentation at Aesthetica MedSpa is a quick, out patient procedure that will create a more youthful looking face. Dr. Sneed uses restylane and juvederm to plump up your lips. He may recommend combing this lip enhancement procedure with injections of radiesse to re volumize your facial features. To learn more about this procedure, visit http://www.amedspa.com/austin-lip-injections.php.

Austin cosmetic dermatologist Dr. David Sneed guides us through one of his Austin lip augmentation procedures using Juvederm and Radiesse. Lip injections using soft tissue fillers, like Restylane & Juvederm, is a great way to revolumize deflated lips and restore a soft look of youth and vitality to your face. To learn more about lip augmentation in Austin, TX, please visit http://www.amedspa.com/austin-lip-injections.php
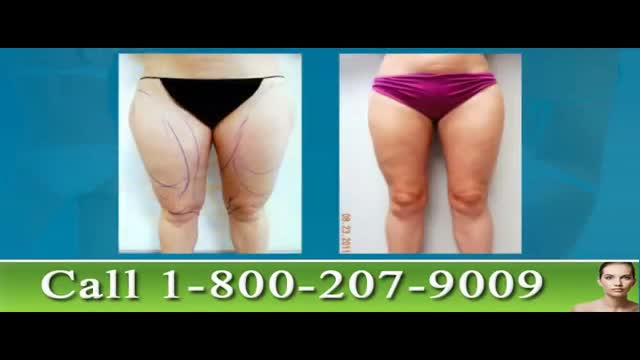
These Tampa Thigh Liposuction before and after photos show the work of renowned liposculpting surgeon Dr. Thomas Su. Dr. Su, of the Artistic Liposculpting Center specializes in body contouring procedures like leg liposuction. Thigh lipo is perhaps one of the most cosmetically rewarding liposuction procedures as it enhances the patient’s overall shape and contour. To learn more about leg and thigh lipo in Tampa, please visit http://www.artlipo.com/leg-liposuction-in-tampa-bay.html

Renowned for her work in reinnervated autologous muscle-preserving perforator flaps, Dr. Aldona Spiegel is one of the top breast reconstruction surgeons in the nation. Dr. Spiegel's office is located in the Houston Methodist Hospital and is affiliated with their Breast Center. These DIEP Flap before and after photos depict some of her recent work. To learn more about DIEP Flap reconstruction in Houston, please visit http://www.breastrestoration.org/diep_siea.php

TV interview with Dr. Mostafa Yakoot, MD discussing latest researches on herbal drugs

Women Health Tips: Importance of getting a pap smear, breast exam, and mammogram
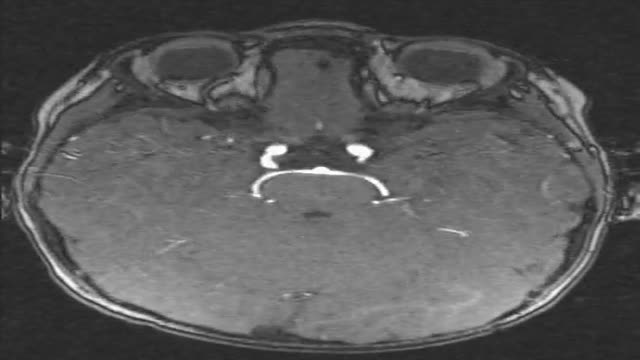
2 year old boy with chronic sinusitis, headache, vertigo problem, decreased vision and hearing. Repeated lung infections. Can you see it? Pay also special attention to the ears.

35 year old women with breathing difficulties for 6 months and feels like fluid is leaking down her front and back. Pain in thorax, lower back and pelvic. Weight loss. Was exposed to mold for a 2 years. Has a dog witch has persistent worm infection. Also been traveling out of the country.

http://www.turkey-ivf.com http://www.tupbebek-istanbul.com We offer a wide range of Assisted Reproductive Technologies including IVF and ICSI (Intracytoplasmic Sperm Injection), Embryo and Sperm Cryopresevation, Intrauterine inseminations (IUI), ovulation induction, Co-Culture (Artificial Uterus). Also other micromanipulation methods (assisted hatching, defragmentation, blastomer biopsy), epididymal or testicular sperm aspiration/extraction (PESA, TESA, TESE or micro TESE) are carried out in our laboratory.
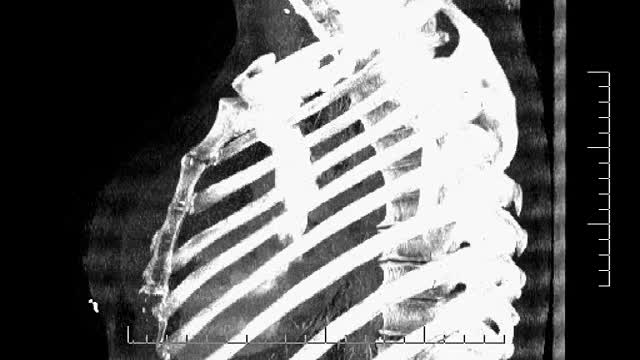
35 year old women with breathing difficulties for 6 months and feels like fluid is leaking down her front and back. Was exposed to mold for a 2 years. Has a dog witch has persistent worm infection. Breast implants 10 years ago.
TV interview with Dr. Mostafa Yakoot, MD discussing his recently published study for efficacy of lettuce seed oil in patients with insomnia

TV interview with Dr. Mostafa Yakoot, MD discussing the published study for efficacy of a new local cream for diabetic foot ulcers. دكتور مصطفى ياقوت علاج جديد للقدم السكرى

Respiratory System Structure and Functions

The Respiratory System
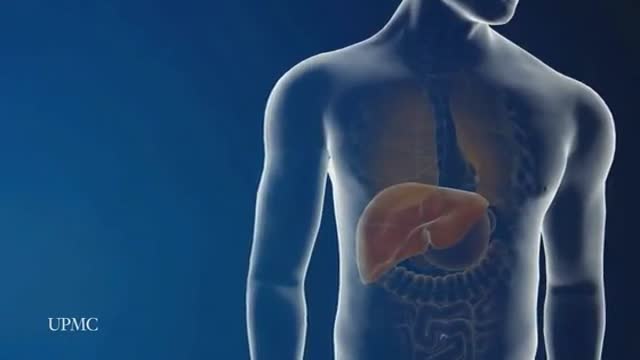
Laparoscopic Liver Surgery 3D Animation

Davinci Robotic Prostatectomy Animation

How Alcohol Affects Your Body
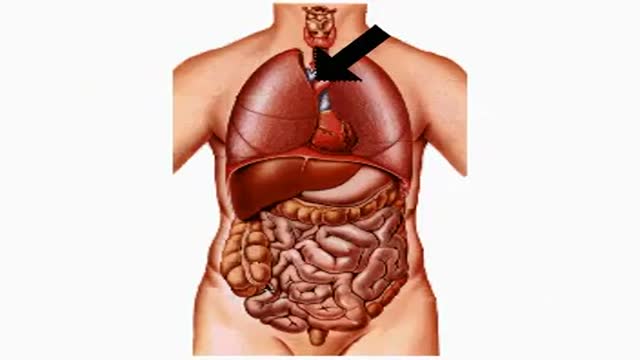
How Smoking and Drinking Affect Your Body
