- Physical Examination
- Surgical Examination
- Ophthalmology
- Clinical Skills
- Orthopedics
- Surgery Videos
- Laparoscopy
- Pediatrics
- Funny Videos
- Cardiothoracic Surgery
- Nursing Videos
- Plastic Surgery
- Otorhinolaryngology
- Histology and Histopathology
- Neurosurgery
- Dermatology
- Pediatric Surgery
- Urology
- Dentistry
- Oncology and Cancers
- Anatomy Videos
- Health and Fitness
- Radiology
- Anaesthesia
- Physical Therapy
- Pharmacology
- Interventional Radiology
- Cardiology
- Endocrinology
- Gynecology
- Emergency Medicine
- Psychiatry and Psychology
- Childbirth Videos
- General Medical Videos
- Nephrology
- Physiology
- Diet and Food Health
- Diabetes Mellitus
- Neurology
- Women Health
- Osteoporosis
- Gastroenterology
- Pulmonology
- Hematology
- Rheumatology
- Toxicology
- Nuclear Medicine
- Infectious Diseases
- Vascular Disease
- Reproductive Health
- Burns and Wound Healing
- Other
Latest videos
Liver Cancer 3D Animation
DrPhil
1,998 Views • 2 years ago
Liver Cancer 3D Animation

STOP SMOKING
DrPhil
10,594 Views • 2 years ago
STOP SMOKING
alisklamp child circumcision
ozzy_tr
9,661 Views • 2 years ago
this video shows how the child circumcision is easy and safe with alisklamp

alisklamp in Africa
ozzy_tr
4,440 Views • 2 years ago
this video shows how the adult circumcision is easy by the alisklamp

Cell Organelles in 3D
DrPhil
8,915 Views • 2 years ago
Cell Organelles in 3D

The Cardiac Cycle
DrPhil
15,081 Views • 2 years ago
The Cardiac Cycle
The Heart How It Works
DrPhil
10,090 Views • 2 years ago
The Heart How It Works
3D Heart Attack
DrPhil
24,247 Views • 2 years ago
3D Heart Attack
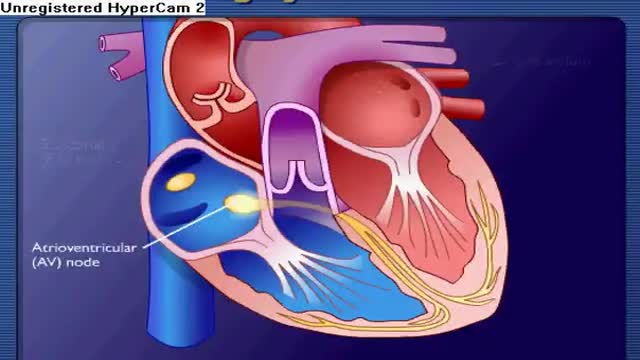
Conductive System of the Heart
DrPhil
7,244 Views • 2 years ago
Conductive System of the Heart
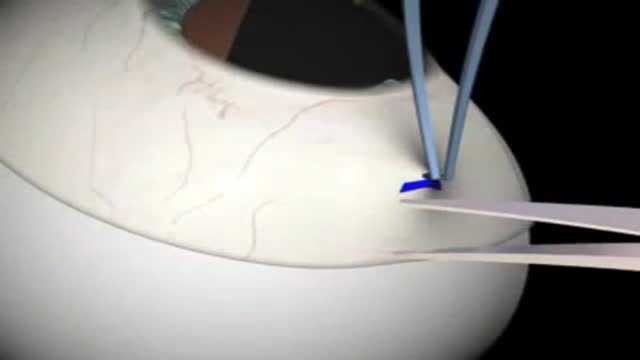
Glaucoma Surgery 3D Animation
DrPhil
7,447 Views • 2 years ago
Glaucoma Surgery 3D Animation
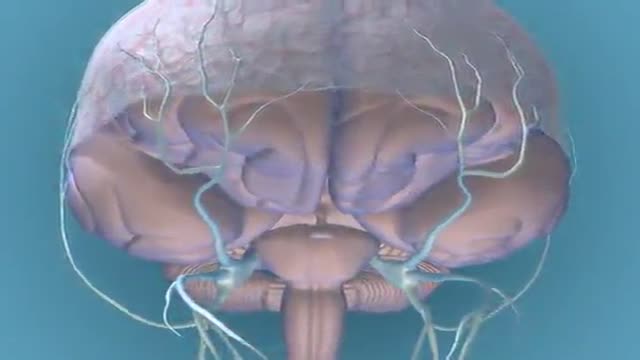
Migraine
DrPhil
18,823 Views • 2 years ago
Migraine
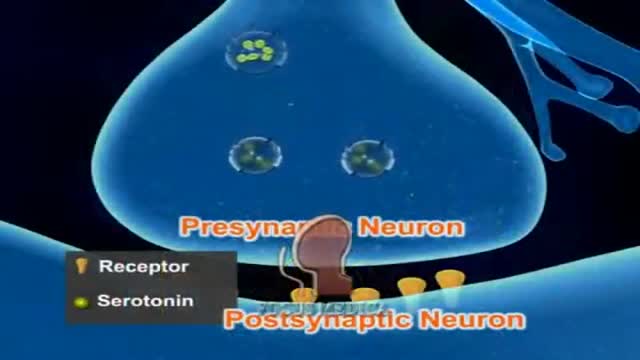
Migraine Pathophysiology 3D Animation
DrPhil
28,358 Views • 2 years ago
Migraine Pathophysiology 3D Animation
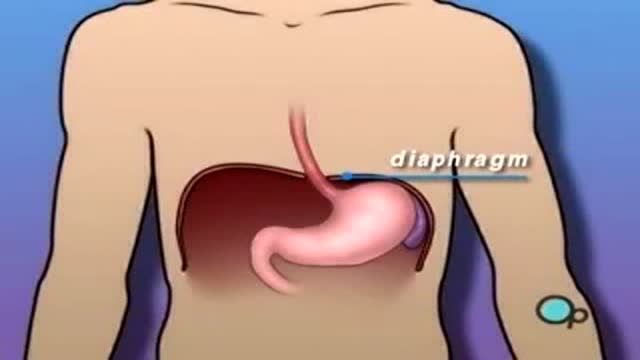
Hiatal Hernia 3D Medical Animation
DrPhil
8,811 Views • 2 years ago
Hiatal Hernia 3D Medical Animation
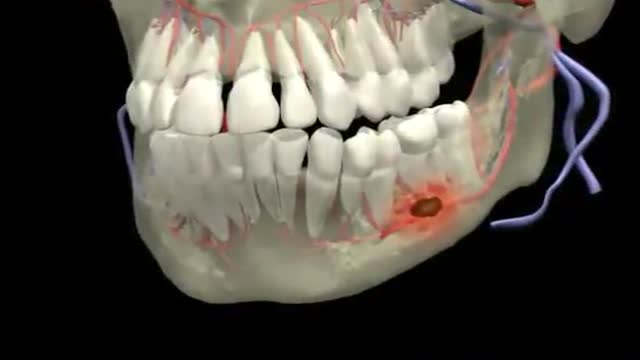
Dental Abscess 3D Animation
Scott
8,677 Views • 2 years ago
Dental Abscess 3D Animation

Don’t be left in the Dark
News Canada
12,420 Views • 2 years ago
Understanding narcolepsy symptoms to improve alertness.

Emergency Medicine Importance
Scott
9,204 Views • 2 years ago
Emergency Medicine Importance If London Was Occupied

Ventricular Assist Devices
Scott
7,369 Views • 2 years ago
Ventricular Assist Devices

Mechanical Circulatory Support
Scott
6,294 Views • 2 years ago
Mechanical Circulatory Support
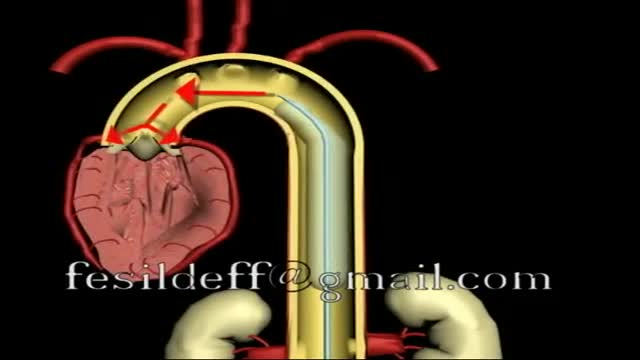
Intra Aortic Balloon Pump
Scott
15,754 Views • 2 years ago
Intra Aortic Balloon Pump
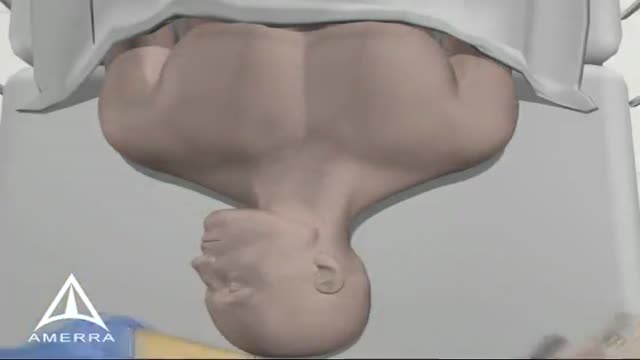
Central Line Placement 3D Animation
Scott
1,598 Views • 2 years ago
Central Line Placement 3D Animation
Showing 260 out of 261
