- Physical Examination
- Surgical Examination
- Ophthalmology
- Clinical Skills
- Orthopedics
- Surgery Videos
- Laparoscopy
- Pediatrics
- Funny Videos
- Cardiothoracic Surgery
- Nursing Videos
- Plastic Surgery
- Otorhinolaryngology
- Histology and Histopathology
- Neurosurgery
- Dermatology
- Pediatric Surgery
- Urology
- Dentistry
- Oncology and Cancers
- Anatomy Videos
- Health and Fitness
- Radiology
- Anaesthesia
- Physical Therapy
- Pharmacology
- Interventional Radiology
- Cardiology
- Endocrinology
- Gynecology
- Emergency Medicine
- Psychiatry and Psychology
- Childbirth Videos
- General Medical Videos
- Nephrology
- Physiology
- Diet and Food Health
- Diabetes Mellitus
- Neurology
- Women Health
- Osteoporosis
- Gastroenterology
- Pulmonology
- Hematology
- Rheumatology
- Toxicology
- Nuclear Medicine
- Infectious Diseases
- Vascular Disease
- Reproductive Health
- Burns and Wound Healing
- Other
Latest videos
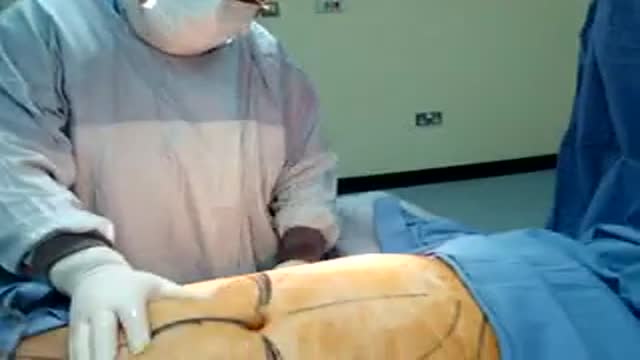
DERMOLIPECTOMY-QATAR DUBAI

Islam and Medicine

Therapeutic Interaction

Nursing A Touch Worthes 1000 Words

Problem Behaviors

Time Management and Work Organization

Staff Communication and Interaction

Creating polidocanol foam

AL EMADI HOSPITAL-QATAR-DOHA AMERICAN BOARD CERTIFICATE AESTHETIC MEDICINE 0097455742973

AL EMADI HOSPITAL-QATAR-DOHA AMERICAN BOARD CERTIFICATE AESTHETIC MEDICINE drkhsh2001@yahoo.com http://www.kamalsaleh.sptechs.com
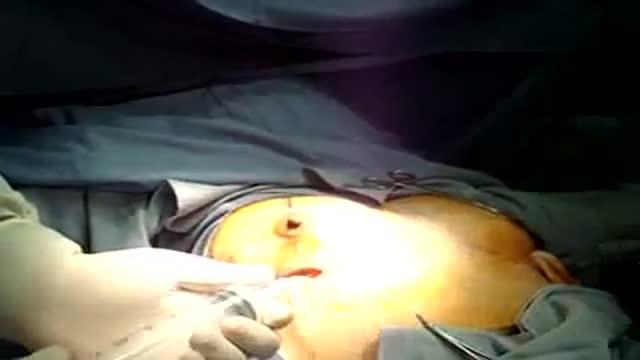
breast implants-breast surgery
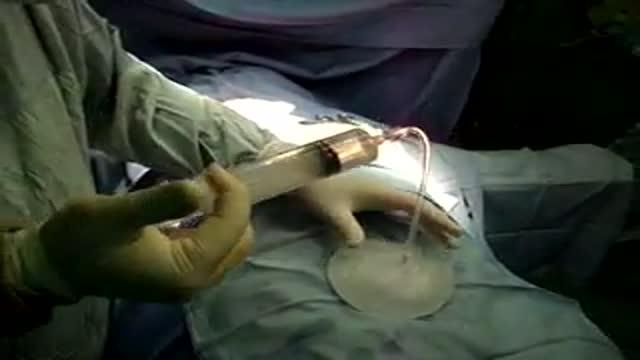
breast augmentation-breast implants

qatar liposuction clinics
AL EMADI HOSPITAL-QATAR-DOHA AMERICAN BOARD CERTIFICATE AESTHETIC MEDICINE 0097455742973 drkhsh2001@yahoo.com http://www.kamalsaleh.sptechs.com

Venepuncture Tutorial HD Drawing Blood
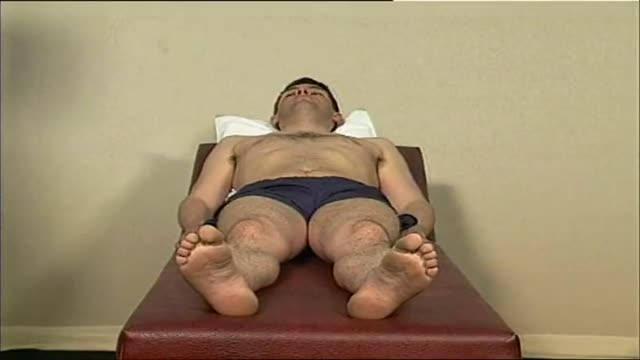
Hip Exam Video
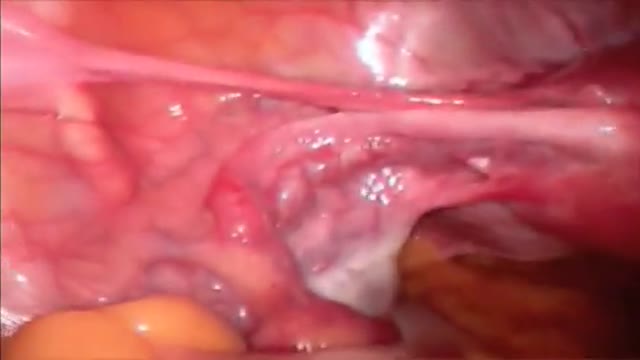
Total Hysterectomy Laparoscopic HD

Endometrial Biopsy of Uterus

Large Bowel Epiploica Laparoscopic Resection
Laparoscopic Uterosacral Colpoplexy HD
