- Physical Examination
- Surgical Examination
- Ophthalmology
- Clinical Skills
- Orthopedics
- Surgery Videos
- Laparoscopy
- Pediatrics
- Funny Videos
- Cardiothoracic Surgery
- Nursing Videos
- Plastic Surgery
- Otorhinolaryngology
- Histology and Histopathology
- Neurosurgery
- Dermatology
- Pediatric Surgery
- Urology
- Dentistry
- Oncology and Cancers
- Anatomy Videos
- Health and Fitness
- Radiology
- Anaesthesia
- Physical Therapy
- Pharmacology
- Interventional Radiology
- Cardiology
- Endocrinology
- Gynecology
- Emergency Medicine
- Psychiatry and Psychology
- Childbirth Videos
- General Medical Videos
- Nephrology
- Physiology
- Diet and Food Health
- Diabetes Mellitus
- Neurology
- Women Health
- Osteoporosis
- Gastroenterology
- Pulmonology
- Hematology
- Rheumatology
- Toxicology
- Nuclear Medicine
- Infectious Diseases
- Vascular Disease
- Reproductive Health
- Burns and Wound Healing
- Other
Top videos
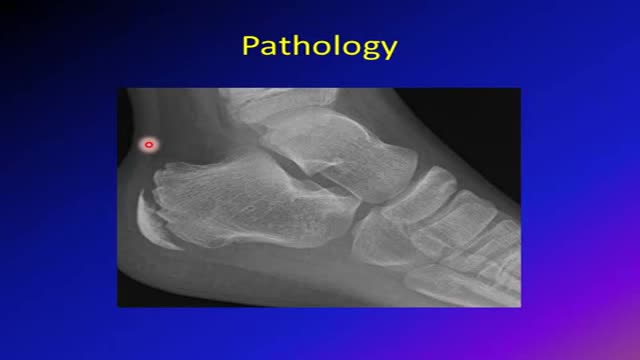
Sever's disease (also known as calcaneal apophysitis) is a type of bone injury in which the growth plate in the lower back of the heel, where the Achilles tendon (the heel cord that attaches to the growth plate) attaches, becomes inflamed and causes pain.
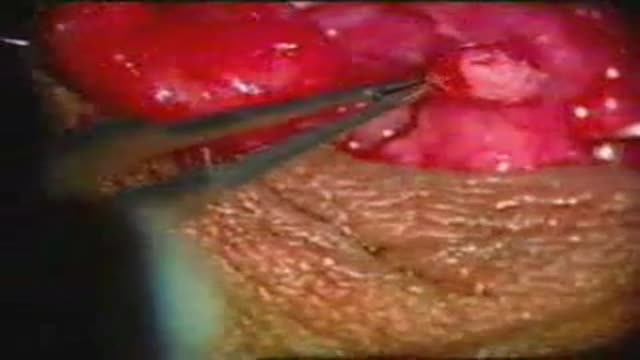
The operation for reversal of vasectomy

Menorrhagia is the medical term for menstrual periods with abnormally heavy or prolonged bleeding. Although heavy menstrual bleeding is a common concern, most women don't experience blood loss severe enough to be defined as menorrhagia.
What are blackheads? Blackheads are small bumps that appear on your skin due to clogged hair follicles. These bumps are called blackheads because the surface looks dark or black. Blackheads are a mild type of acne that usually form on the face, but they can also appear on the following body parts: back chest neck arms shoulders Acne affects nearly 50 million Americans and is the most common skin disorder in the United States, according to the American Academy of Dermatology. What do blackheads look like? What causes blackheads? Blackheads form when a clog or plug develops in the opening of hair follicles in your skin. Each follicle contains one hair and a sebaceous gland that produces oil. This oil, called sebum, helps keep your skin soft. Dead skin cells and oils collect in the opening to the skin follicle, producing a bump called a comedo. If the skin over the bump stays closed, the bump is called a whitehead. When the skin over the bump opens, exposure to the air causes it to look black and a blackhead forms. Some factors can increase your chances of developing acne and blackheads, including: producing too much body oil the buildup of the Propionibacterium acnes bacteria on the skin irritation of the hair follicles when dead skins cells don’t shed on a regular basis undergoing hormonal changes that cause an increase in oil production during the teen years, during menstruation, or while taking birth control pills taking certain drugs, such as corticosteroids, lithium, or androgens Some people believe that what you eat or drink can affect acne. Dairy products and foods that increase blood sugar levels, such as carbohydrates, may play a part in triggering acne, but researchers aren’t convinced that there’s a strong connection. ADVERTISING What are symptoms of blackheads? Because of their dark color, blackheads are easy to spot on the skin. They’re slightly raised, although they aren’t painful because they aren’t inflamed like pimples. Pimples form when bacteria invade the blockage in the hair follicle, causing redness and inflammation. How are blackheads treated? Over-the-counter (OTC) treatments Many acne medications are available at drug and grocery stores and online without a prescription. These medications are available in cream, gel, and pad form and are put directly on your skin. The drugs contain ingredients such as salicylic acid, benzoyl peroxide, and resorcinol. They work by killing bacteria, drying excess oil, and forcing the skin to shed dead skin cells. Prescription medications If OTC treatment doesn’t improve your acne, your doctor may suggest that you use stronger prescription medications. Medications that contain vitamin A keep plugs from forming in the hair follicles and promote more rapid turnover of skin cells. These medications are applied directly to your skin and can include tretinoin, tazarotene, or adapalene. Your doctor may also prescribe another type of topical medication that contains benzoyl peroxide and antibiotics. If you have pimples or acne cysts in addition to your blackheads, this type of medication may be particularly helpful. Manual removal Dermatologists or specially trained skin care professionals use a special instrument called a round loop extractor to remove the plug causing the blackhead. After a small opening is made in the plug, the doctor applies pressure with the extractor to remove the clog. Microdermabrasion During microdermabrasion, a doctor or skin care professional uses a special instrument that contains a rough surface to sand the top layers of your skin. Sanding the skin removes clogs that cause blackheads. Chemical peels Chemical peels also remove clogs and get rid of the dead skins cells that contribute to blackheads. During a peel, a strong chemical solution is applied to the skin. Over time, the top layers of the skin peel off, revealing smoother skin underneath. Mild peels are available over the counter, while stronger peels are performed by dermatologists or other skincare professionals. Laser and light therapy Laser and light therapies use tiny beams of intense light to decrease oil production or kill bacteria. Both lasers and light beams reach below the surface of the skin to treat blackheads and acne without damaging the top layers of the skin. How can blackheads be prevented? You can prevent blackheads without spending a lot of money by trying a few of the following ideas: Wash regularly Wash your face when you wake up and before you go to bed to remove oil buildup. Washing more than twice each day can irritate your skin and make your acne worse. Use a gentle cleanser that doesn’t make your skin red or irritated. Some acne cleansing products have antibacterial ingredients that kill P. acnes bacteria. Consider washing your hair every day, too, particularly if it’s oily. Hair oils can contribute to clogged pores. It’s also important to wash your face after you eat oily foods such as pizza, because oil from these foods can clog pores. Use oil-free products Any product that contains oil can contribute to new blackheads. Choose oil-free or noncomedogenic makeup, lotions, and sunscreens to avoid making your problem worse. Try an exfoliating product Exfoliating scrubs and masks remove dead skin cells from your face and can help reduce blackheads. Look for products that don’t irritate your skin.


Supraventricular tachycardia (SVT) is an abnormal condition of heart which increases normal heartbeat rate rapidly. Normally, heartbeat rate should be between 80 to 100 beats per minute. For more info: https://goo.gl/14btbU
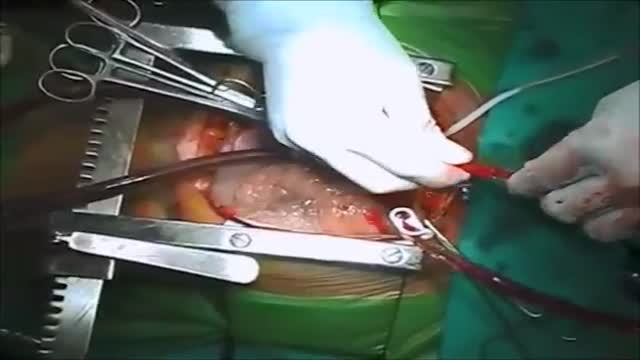
Ventricular septal rupture (VSR) is a rare but lethal complication of myocardial infarction (MI). The event occurs 2-8 days after an infarction and often precipitates cardiogenic shock. [1] The differential diagnosis of postinfarction cardiogenic shock should exclude free ventricular wall rupture and rupture of the papillary muscles. (See the image below.)

Cobalamin (vitamin B12) deficiency is particularly common in the elderly (>65 years of age), but is often unrecognized because of its subtle clinical manifestations; although they can be potentially serious, particularly from a neuropsychiatric and hematological perspective.

The surgeon may use treatment options such as thoracoscopy, electrocautery, laser treatment, resection of blebs or pleura, or open thoracotomy. Other surgical indications are as follows: Persistent air leak for longer than 7 days. Recurrent, ipsilateral pneumothorax.
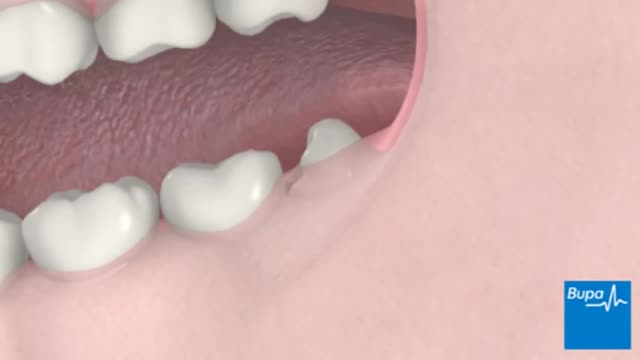
A wisdom tooth or third molar is one of the three molars per quadrant of the human dentition. It is the most posterior of the three. Wisdom teeth generally erupt between the ages of 17
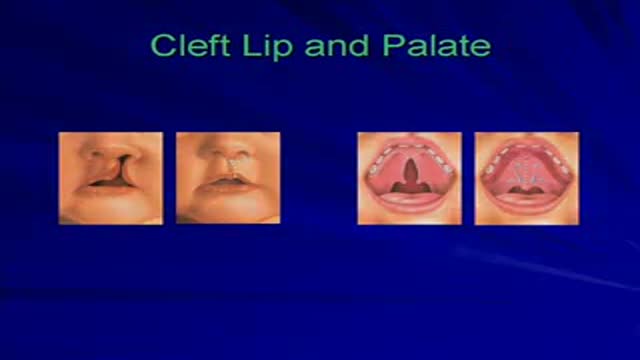
This video is a collection of selected cases of Plastic Surgery performed on children with congenital deformities.
Errata: Cleft Lip Case 2 has a center photo which belongs to case 1 at day of surgery.

Vaginismus Pain Management

Scrubbing of Surgical Hands

Sepsis is a potentially life-threatening complication of an infection. Sepsis occurs when chemicals released into the bloodstream to fight the infection trigger inflammatory responses throughout the body. This inflammation can trigger a cascade of changes that can damage multiple organ systems, causing them to fail. If sepsis progresses to septic shock, blood pressure drops dramatically, which may lead to death. Anyone can develop sepsis, but it's most common and most dangerous in older adults or those with weakened immune systems. Early treatment of sepsis, usually with antibiotics and large amounts of intravenous fluids, improves chances for survival. Symptoms & causes Symptoms Many doctors view sepsis as a three-stage syndrome, starting with sepsis and progressing through severe sepsis to septic shock. The goal is to treat sepsis during its early stage, before it becomes more dangerous. Sepsis To be diagnosed with sepsis, you must exhibit at least two of the following symptoms, plus a probable or confirmed infection: Body temperature above 101 F (38.3 C) or below 96.8 F (36 C) Heart rate higher than 90 beats a minute Respiratory rate higher than 20 breaths a minute Severe sepsis Your diagnosis will be upgraded to severe sepsis if you also exhibit at least one of the following signs and symptoms, which indicate an organ may be failing: Significantly decreased urine output Abrupt change in mental status Decrease in platelet count Difficulty breathing Abnormal heart pumping function Abdominal pain Septic shock To be diagnosed with septic shock, you must have the signs and symptoms of severe sepsis — plus extremely low blood pressure that doesn't adequately respond to simple fluid replacement. When to see a doctor Most often sepsis occurs in people who are hospitalized. People in the intensive care unit are especially vulnerable to developing infections, which can then lead to sepsis. If you get an infection or if you develop signs and symptoms of sepsis after surgery, hospitalization or an infection, seek medical care immediately. Causes While any type of infection — bacterial, viral or fungal — can lead to sepsis, the most likely varieties include: Pneumonia Abdominal infection Kidney infection Bloodstream infection (bacteremia) The incidence of sepsis appears to be increasing in the United States. The causes of this increase may include: Aging population. Americans are living longer, which is swelling the ranks of the highest risk age group — people older than 65. Drug-resistant bacteria. Many types of bacteria can resist the effects of antibiotics that once killed them. These antibiotic-resistant bacteria are often the root cause of the infections that trigger sepsis. Weakened immune systems. More Americans are living with weakened immune systems, caused by HIV, cancer treatments or transplant drugs. Risk factors Sepsis is more common and more dangerous if you: Are very young or very old Have a compromised immune system Are already very sick, often in a hospital's intensive care unit Have wounds or injuries, such as burns Have invasive devices, such as intravenous catheters or breathing tubes Complications Sepsis ranges from less to more severe. As sepsis worsens, blood flow to vital organs, such as your brain, heart and kidneys, becomes impaired. Sepsis can also cause blood clots to form in your organs and in your arms, legs, fingers and toes — leading to varying degrees of organ failure and tissue death (gangrene). Most people recover from mild sepsis, but the mortality rate for septic shock is nearly 50 percent. Also, an episode of severe sepsis may place you at higher risk of future infections.

Transvenous cardiac pace maker, also called endocardial pacing, is a potentially life saving intervention used primarily to correct profound bradycardia. It can be used to treat symptomatic bradycardias that do not respond to transcutaneous pacing or to drug therapy.
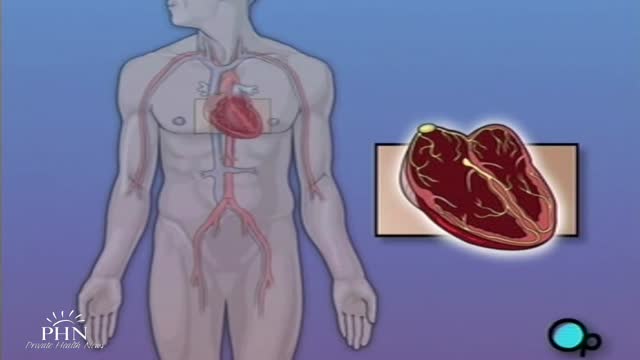
Permanent pacemaker insertion is considered a minimally invasive procedure. Transvenous access to the heart chambers under local anesthesia is the favored technique, most commonly via the subclavian vein, the cephalic vein, or (rarely) the internal jugular vein or the femoral vein.
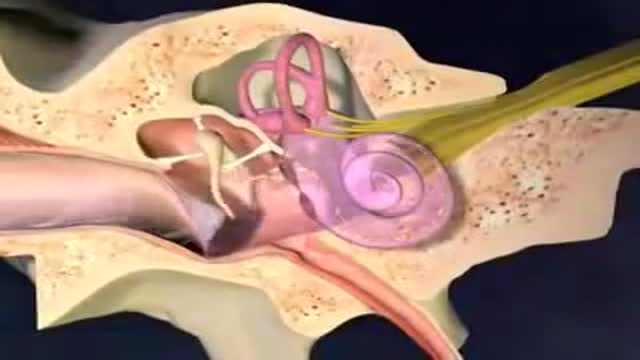
Acute otitis media: Inflammation of the middle ear in which there is fluid in the middle ear accompanied by signs or symptoms of ear infection: a bulging eardrum usually accompanied by pain; or a perforated eardrum, often with drainage of purulent material (pus).

Official Ninja Nerd Website: https://ninjanerd.org
You can find the NOTES and ILLUSTRATIONS for this lecture on our website at:
https://www.ninjanerd.org/lect....ure/hemodialysis-ncl
Ninja Nerds!
In this lecture Professor Kristin Beach, MSN, BSN, RN will be discussing Hemodialysis. Hemodialysis is a procedure where a dialysis machine and a special filter called an artificial kidney, or a dialyzer, are used to clean your blood. In order to receive dialysis, a minor surgery is performed to the arm to create an arteriovenous shunt.
Table of Contents:
0:00 Lab
0:07 Hemodialysis Introduction
0:27 Defining Hemodialysis
11:02 Dialysis: Pre-Procedure
14:52 Dialysis: Intra-Procedure
17:34 Dialysis: Post-Procedure
18:58 Complications
27:35 Comment, Like, SUBSCRIBE!
APPAREL |
https://www.amazon.com/s?k=ninja+nerd&ref=nb_sb_noss_2
PODCAST |
Apple Podcast: https://podcasts.apple.com/us/....podcast/ninja-nerd/i
Spotify: https://open.spotify.com/show/....2ZDXoakATwCgkRH3EpCZ
Google Podcast: https://podcasts.google.com/fe....ed/aHR0cHM6Ly9mZWVkc
DONATE
PAYPAL | https://www.paypal.com/paypalme/ninjanerdscience
SOCIAL MEDIA
FACEBOOK | https://www.facebook.com/NinjaNerdlectures
INSTAGRAM | https://www.instagram.com/ninjanerdlectures
TWITTER | https://twitter.com/ninjanerdsci
@NinjaNerdSci
DISCORD | https://discord.gg/3srTG4dngW
#ninjanerd #hemodialysis #nursing

Smead Jones Sutures - Far Far- Near Near
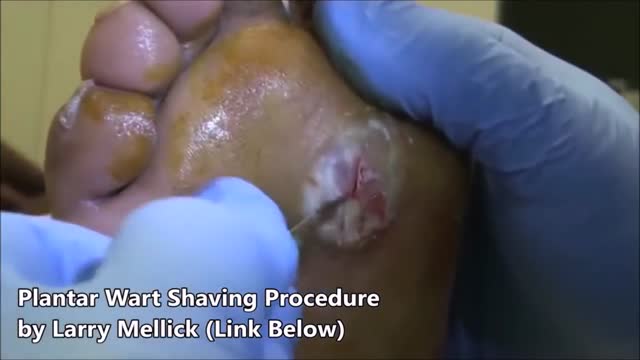
Plantar warts are hard, grainy growths that usually appear on the heels or balls of your feet, areas that feel the most pressure. This pressure also may cause plantar warts to grow inward beneath a hard, thick layer of skin (callus). Plantar warts are caused by the human papillomavirus (HPV). The virus enters your body through tiny cuts, breaks or other weak spots on the bottom of your feet. Most plantar warts aren't a serious health concern and may not require treatment. But plantar warts can cause discomfort or pain. If self-care treatments for plantar warts don't work, you may want to see your doctor to have them removed.
