- Physical Examination
- Surgical Examination
- Ophthalmology
- Clinical Skills
- Orthopedics
- Surgery Videos
- Laparoscopy
- Pediatrics
- Funny Videos
- Cardiothoracic Surgery
- Nursing Videos
- Plastic Surgery
- Otorhinolaryngology
- Histology and Histopathology
- Neurosurgery
- Dermatology
- Pediatric Surgery
- Urology
- Dentistry
- Oncology and Cancers
- Anatomy Videos
- Health and Fitness
- Radiology
- Anaesthesia
- Physical Therapy
- Pharmacology
- Interventional Radiology
- Cardiology
- Endocrinology
- Gynecology
- Emergency Medicine
- Psychiatry and Psychology
- Childbirth Videos
- General Medical Videos
- Nephrology
- Physiology
- Diet and Food Health
- Diabetes Mellitus
- Neurology
- Women Health
- Osteoporosis
- Gastroenterology
- Pulmonology
- Hematology
- Rheumatology
- Toxicology
- Nuclear Medicine
- Infectious Diseases
- Vascular Disease
- Reproductive Health
- Burns and Wound Healing
- Other
Top videos

Motor examination of Lower Limb from the USMLE collection

Minimally invasive kidney and ureteral stone surgery using holmium laser performed at El Camino Urology Medical Group,
Aneurysm of Splenic Artery from Cairo College of Medicine Hospitals

Intercostal Nerve Block
Transoral Access in Endoscopic Thyroid Surgery Background: The number of patients demanding endoscopic neck surgery is rising. The access trauma of the axillary, breast and chest approaches is bigger than in open or video assisted surgery. We tested the feasibility of he sublingual transoral access which is in our opinion the only real minimally...-invasive extracollar endoscopic access to the thyroid gland Methods: We performed an experimental investigation in a porcine model. In 10 pigs we made 10 endoscopic transoral thyroidectomys with a modified axilloscope with the help of ultrasonic scissors and a neuro-monitoring system for identification of the recurrent laryngeal nerve. Results: The average operation time from the introduction to the removal of the obturator just above the larynx was 57 seconds. The mean operation time was 43 minutes. With the help of the neuro-monitoring system we proved in all cases the function of the recurrent laryngeal nerve on both sides. The pigs were observed for another two hours after operation. During and after the operation no complications appeared. Conclusions: We could show that the endoscopic transoral thyroid resection in pigs is possible and save. Our results might be useful for using this access for endoscopic thyroid resection in humans.
The essential steps of a translaminaterminalis approach for removal of craniopharyngiomas

Lap Band Procedure done on a patient with a BMI of 45. Minimal editing and includes narration.

Choking Child Video Demonstration

DMC pain management Specialists Drs. Renee Baugh and Mohamed Othman work to help patients manage and minimize pain, and restore a more satisfying lifestyle. ~ Detroit Medical Center
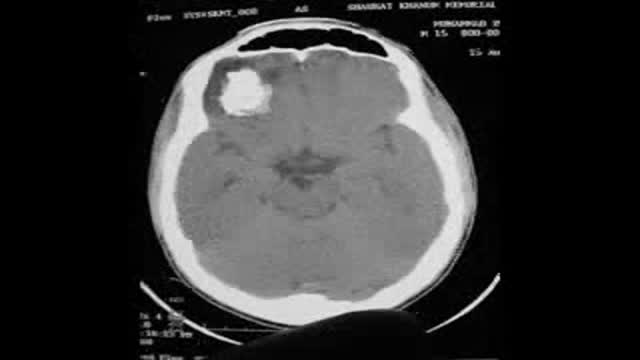
Calcified Brain Abcess complete removal,
surgery

Pediatric Abdomen - Radiology & Imaging Powerpoint Video Lecture presentation with interesting case collection
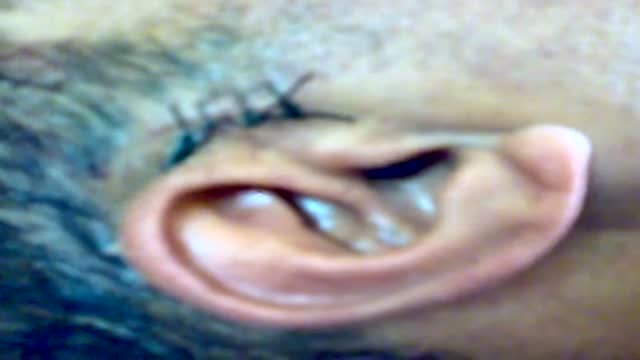
Simple surgery under a local anesthesia can help cluster headaches patients:
www.alisultaneh.8m.com
www.migrainesurgery.4t.com
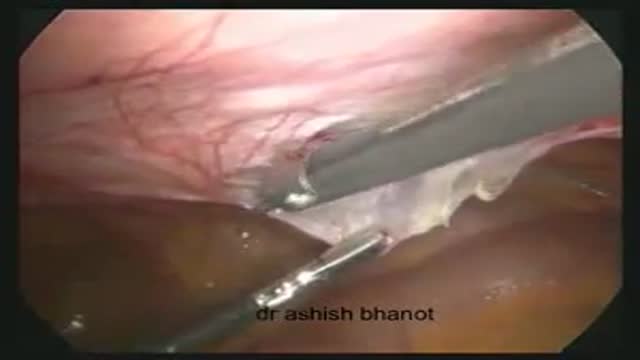
This video is showing the Laparoscopic transabdominal preperitoneal hernia repair for direct inguinal Hernia
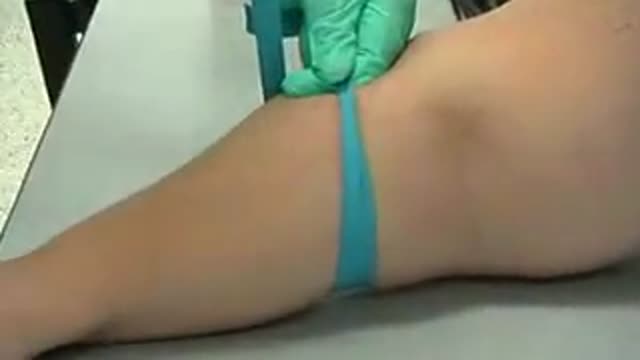
How to properly apply a tourniquet
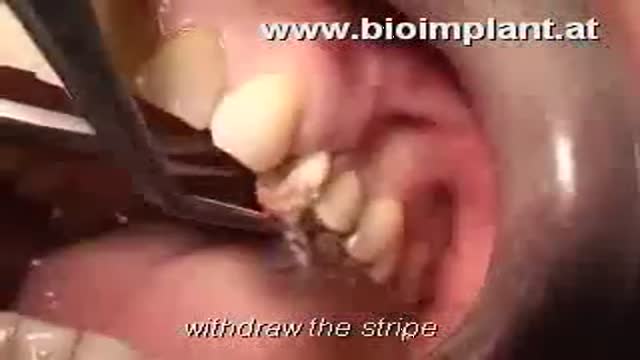
WORLD'S FIRST IMMEDIATE ROOT-ANALOG ZIRCONIA DENTAL IMPLANT amazing video

the video will describe oxyhemoglobin dissociation curve. please see my website for disclaimer.
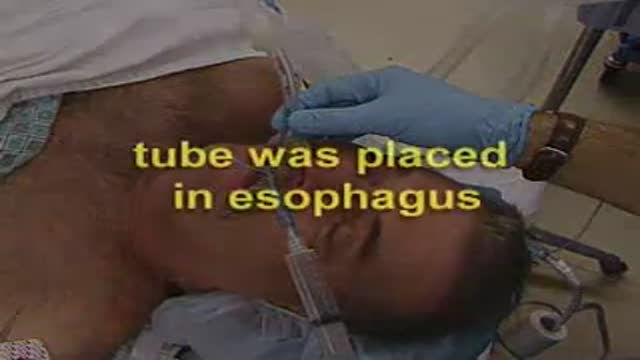
A video showing Intubation of the Esophagus
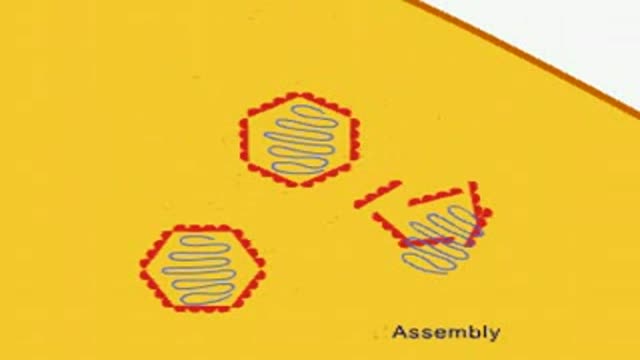
Showing how viruses multiply in general

In this video Erin K, a tubal reversal patient, explains the symptoms she experienced while suffering from Post Tubal Ligation Syndrome (PTLS). After having tubal reversal surgery her symptoms were relieved. Although numerous women suffer from adverse symptoms after having a tubal ligation, many physicians do not believe PTLS exists. In an ongoing study of over 300 patients reporting Post Tubal Ligation symptoms more than 90% have found relief after tubal reversal at Chapel Hill Tubal Reversal Center.

Bruce had a stroke 2 years ago. Then, he broke his hip. The stroke affected his right side, and he has had limited mobility and was using a walker for recovery, Bruce could only walk with breaks and was hunched over. The GlideTrak opened Bruce's posture and allowed him to breath better and allowed for over 25 minutes of walking exercise, greatly increasing the Patients self-confidence and at the end he was actually able to stand on his own feet, with the straps as guides, whereas this was not possible before his sessions on the GlideTrak. clean, water-damp cloth. Repeat application procedure as needed.
