- Physical Examination
- Surgical Examination
- Ophthalmology
- Clinical Skills
- Orthopedics
- Surgery Videos
- Laparoscopy
- Pediatrics
- Funny Videos
- Cardiothoracic Surgery
- Nursing Videos
- Plastic Surgery
- Otorhinolaryngology
- Histology and Histopathology
- Neurosurgery
- Dermatology
- Pediatric Surgery
- Urology
- Dentistry
- Oncology and Cancers
- Anatomy Videos
- Health and Fitness
- Radiology
- Anaesthesia
- Physical Therapy
- Pharmacology
- Interventional Radiology
- Cardiology
- Endocrinology
- Gynecology
- Emergency Medicine
- Psychiatry and Psychology
- Childbirth Videos
- General Medical Videos
- Nephrology
- Physiology
- Diet and Food Health
- Diabetes Mellitus
- Neurology
- Women Health
- Osteoporosis
- Gastroenterology
- Pulmonology
- Hematology
- Rheumatology
- Toxicology
- Nuclear Medicine
- Infectious Diseases
- Vascular Disease
- Reproductive Health
- Burns and Wound Healing
- Other
Top videos
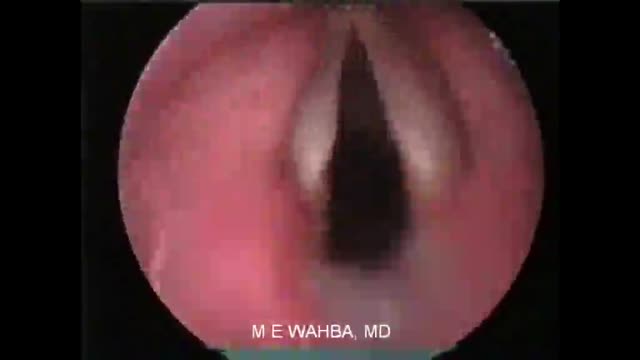
Of course, these vocal cords are not mine, because I am the one who captured this video before I performed surgery for the patient. See how the cords are normal and freely mobile. Talkative persons have something else....imagine
Pregnancy All Weeks HD Animation: From www.MedicalVideos.us Showing the progress and mechanisms of pregnancy in each week.
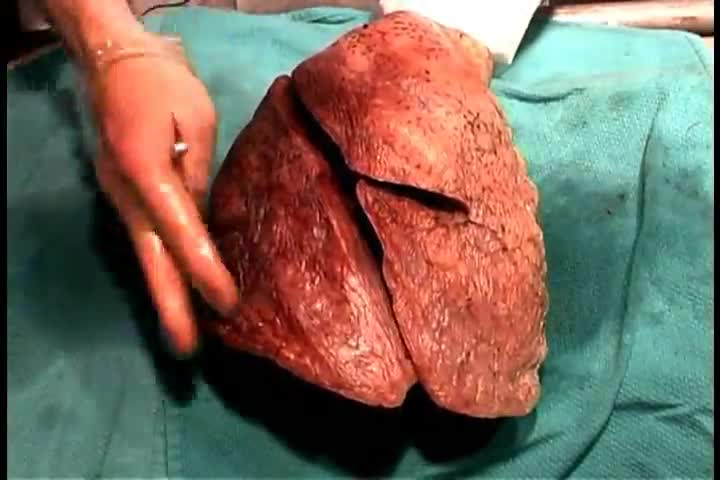
Anatomy of The Anterior Thorax
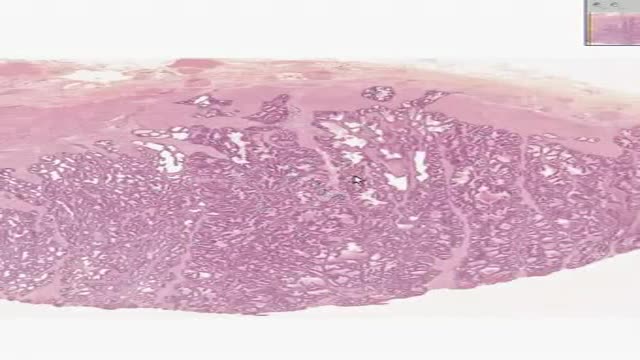
Histology of Prostate
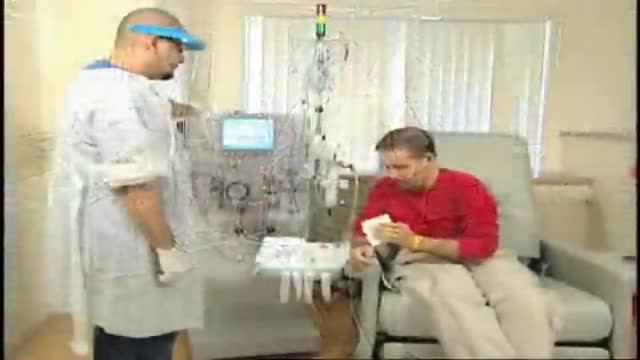
Renal Failure Treatment Options

Mumps Signs Symptoms Complications

Smoking and Breast Feeding
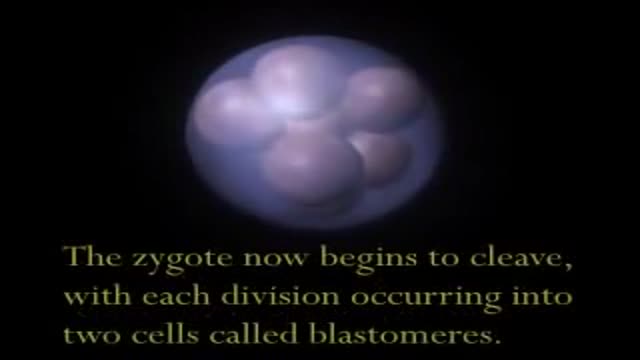
Human Embryo and Fetal Development
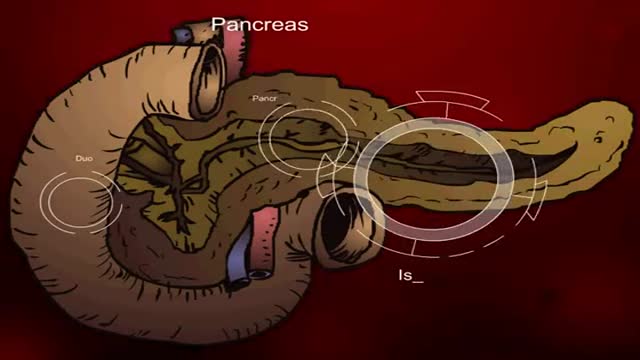
Insulin Processes Mechanism Animation 3D

Total Knee Replacement Patient Information
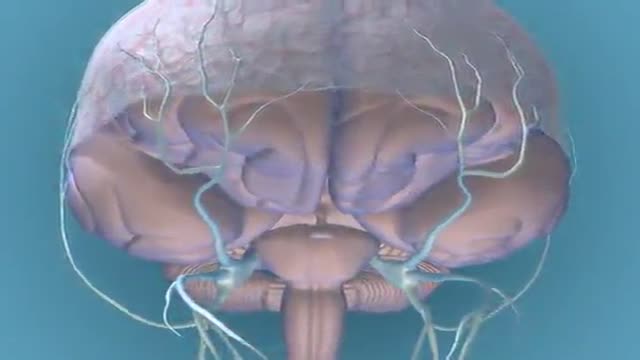
Migraine Pathophysiology 3D Animation

Cell Organelles in 3D
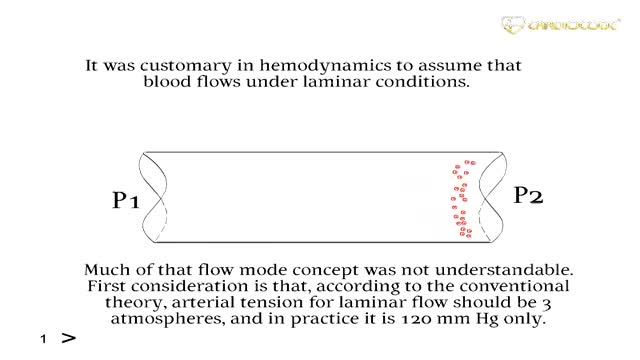
New methods in heart diseases diagnostics and imaging
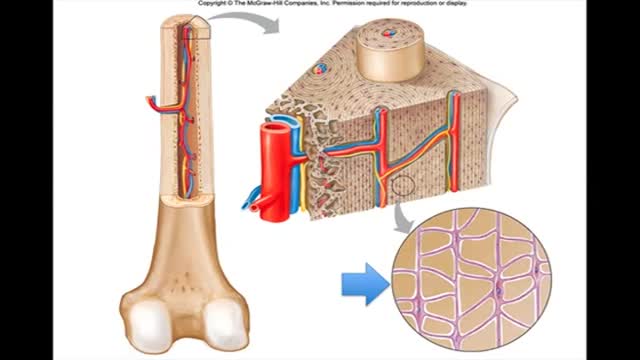
Microscopic Bone Structure

http://www.landging.com/endocrine-system-animation.html
This endocrine system animation demonstrates mechanism of action of human body in 3D.

Understanding Male Infertility
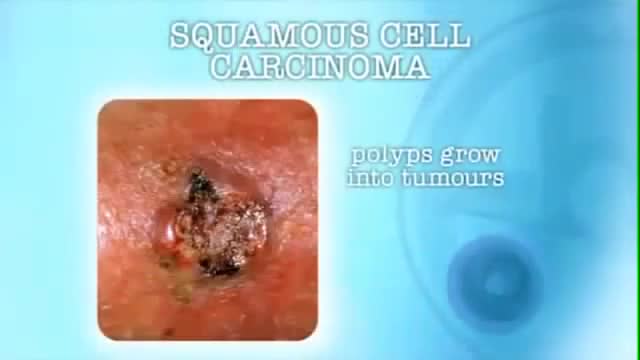
Man had to have aggressive removal of Squamous Cell Carcinoma which resulted in the loss of a large portion of his face.

Tudo Sobre Diabetes, Diabetes Tem Cura, O Que é Diabetes Tipo 2, Plantas Que Curam Diabetes
http://tudo-sobre-diabetes.good-info.co
Cura Naturalmente a Diabetes Tipo 2
A diabetes tipo II se tornou uma das doenças mais comuns nos tempos modernos. A boa notícia é que em pouco menos de um mês, seguindo um plano de alimentação e vida saudável, é possível equilibrar seu nível de açúcar no sangue e prevenir as terríveis consequências que esta doença tem.
A seguir, você encontrará este plano para nivelar o açúcar no sangue e dizer adeus para a diabetes.
Restrinja o consumo de todo o tipo de bebidas.
Realize atividade física de baixo impacto todo o dia, por um mínimo de meia hora.
Elimine por completo de suas refeições, todos os alimentos que contenham farinha branca.
Inclua em sua alimentação habitual, ácidos gordos essenciais (especialmente ácidos ômega 3), inclua também o consumo de frutas secas.
único Sistema Eficiente, Fácil E Natural Para Eliminar Para Sempre O Diabetes. Um Sistema Cientificamente Comprovado
Clique No Link Abaixo Para Verificá-la
http://tudo-sobre-diabetes.good-info.co
Assine O Nosso Canal
https://www.youtube.com/user/dicasdesaude11
https://www.youtube.com/watch?v=61MN7xSR9yA
Tudo Sobre Diabetes, Diabetes Tem Cura, O Que é Diabetes Tipo 2, Plantas Que Curam Diabetes,
diabetes gestacional,
diabetes mellitus tipo 2,
diabetes dieta,
sintomas de diabete,
diabetes tipo 1 e 2,
medicamentos para diabetes,
diabete sintomas,
causas da diabetes,
como evitar diabetes,
sintomas da diabetes tipo 2,
tratamento da diabetes,
o que diabetes,
os sintomas da diabete
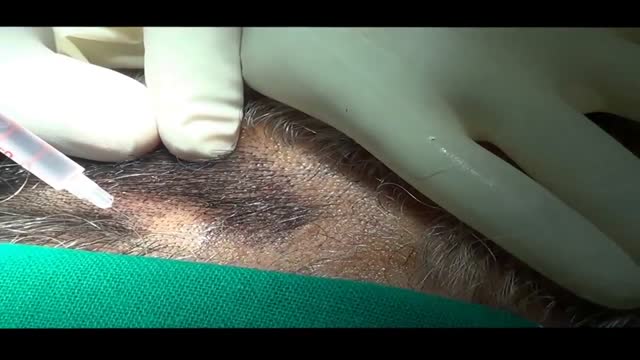
The hair transplant surgeon can accurately estimate the number of follicular grafts that can be obtained from dissecting a donor strip of a given size. The same number of follicular units can be used to cover a specific size bald area regardless of the patient's actual hair density.
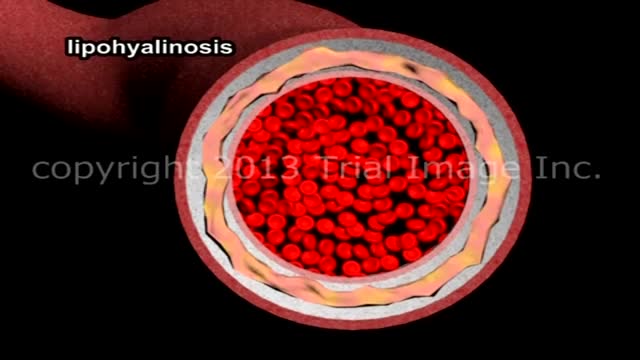
A stroke is a "brain attack". It can happen to anyone at any time. It occurs when blood flow to an area of brain is cut off. When this happens, brain cells are deprived of oxygen and begin to die. When brain cells die during a stroke, abilities controlled by that area of the brain such as memory and muscle control are lost. How a person is affected by their stroke depends on where the stroke occurs in the brain and how much the brain is damaged. For example, someone who had a small stroke may only have minor problems such as temporary weakness of an arm or leg. People who have larger strokes may be permanently paralyzed on one side of their body or lose their ability to speak. Some people recover completely from strokes, but more than 2/3 of survivors will have some type of disability.
