- Physical Examination
- Surgical Examination
- Ophthalmology
- Clinical Skills
- Orthopedics
- Surgery Videos
- Laparoscopy
- Pediatrics
- Funny Videos
- Cardiothoracic Surgery
- Nursing Videos
- Plastic Surgery
- Otorhinolaryngology
- Histology and Histopathology
- Neurosurgery
- Dermatology
- Pediatric Surgery
- Urology
- Dentistry
- Oncology and Cancers
- Anatomy Videos
- Health and Fitness
- Radiology
- Anaesthesia
- Physical Therapy
- Pharmacology
- Interventional Radiology
- Cardiology
- Endocrinology
- Gynecology
- Emergency Medicine
- Psychiatry and Psychology
- Childbirth Videos
- General Medical Videos
- Nephrology
- Physiology
- Diet and Food Health
- Diabetes Mellitus
- Neurology
- Women Health
- Osteoporosis
- Gastroenterology
- Pulmonology
- Hematology
- Rheumatology
- Toxicology
- Nuclear Medicine
- Infectious Diseases
- Vascular Disease
- Reproductive Health
- Burns and Wound Healing
- Other
Top videos
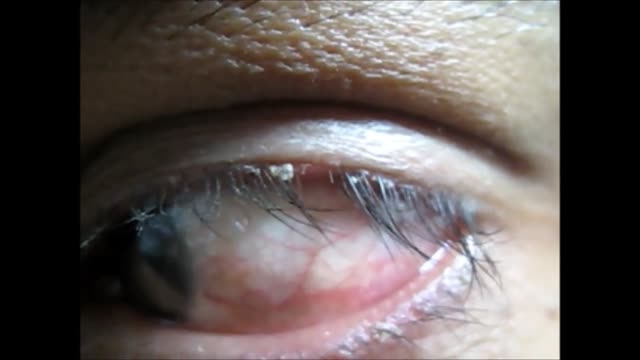
Blepharitis is an inflammation of the eyelids in which they become red, irritated and itchy and dandruff-like scales form on the eyelashes. It is a common eye disorder caused by either bacteria or a skin condition, such as dandruff of the scalp or acne rosacea. It affects people of all ages. Although uncomfortable, blepharitis is not contagious and generally does not cause any permanent damage to eyesight.

To save humanity, a dietitian travels to the past. A lot.
Subscribe now: https://www.youtube.com/c/funn....yordie?sub_confirmat
CREDITS:
Director: Elliot Dickerhoof
Producers: Chuck Armstrong, Charlie Stockman, Elliot Dickerhoof
Writers: Chuck Armstrong & Charlie Stockman
Actors: Chuck Armstrong, Charlie Stockman, Kelly Vrooman
Executive Producer: Darren Miller
DP: Cody Jacobs
Gaffer: Jordan Holtane
AC: Giselle Gonzalez
Sound Mixer: Marcos Castro
Costume Designer: Kate Bergh
Hair and Makeup Artist: Jessica Leigh Schwartz
PA: Elyssa Phillips
Get more Funny Or Die
-------------------------------
Like FOD on Facebook: https://www.facebook.com/funnyordie
Follow FOD on Twitter: https://twitter.com/funnyordie
Follow FOD on Tumblr: http://funnyordie.tumblr.com/
Follow FOD on Instagram: http://instagram.com/funnyordie
Follow FOD on Vine: https://vine.co/funnyordie
Follow FOD on Pinterest: http://www.pinterest.com/funnyordie
Follow FOD on Google+: https://plus.google.com/+funnyordie
See the original at: http://www.funnyordie.com/videos/74dd9afee2
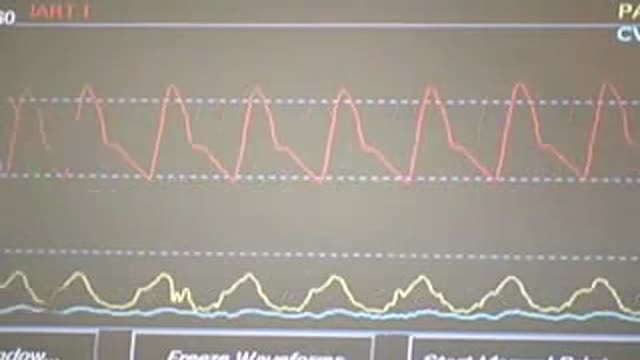
Central Venous Catheter Placement & Pulmonary Artery Catheter Video

Laparoscopy by Dr. Emadi in Qatar
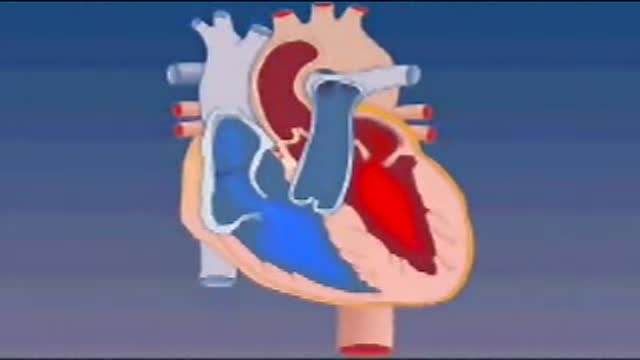
Blood enters the heart through two large veins, the inferior and superior vena cava, emptying oxygen-poor blood from the body into the right atrium. As the atrium contracts, blood flows from your right atrium into your right ventricle through the open tricuspid valve.
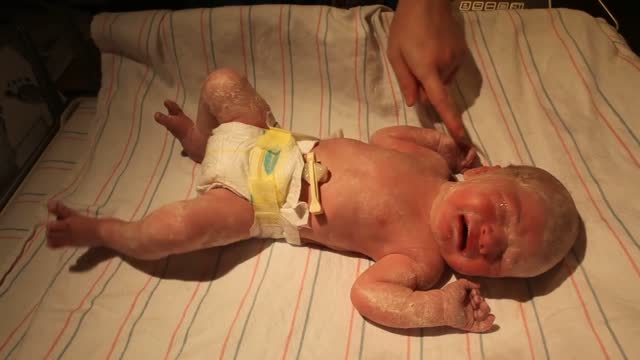
Although the Apgar score was developed in 1952 by an anesthesiologist named Virginia Apgar, you also might hear it referred to as an acronym for: Appearance, Pulse, Grimace, Activity, and Respiration. The Apgar test is usually given to a baby twice: once at 1 minute after birth, and again at 5 minutes after birth.

An abdominal aortic aneurysm is an enlarged area in the lower part of the aorta, the major blood vessel that supplies blood to the body. The aorta, about the thickness of a garden hose, runs from your heart through the center of your chest and abdomen. Because the aorta is the body's main supplier of blood, a ruptured abdominal aortic aneurysm can cause life-threatening bleeding. Depending on the size and the rate at which your abdominal aortic aneurysm is growing, treatment may vary from watchful waiting to emergency surgery. Once an abdominal aortic aneurysm is found, doctors will closely monitor it so that surgery can be planned if it's necessary. Emergency surgery for a ruptured abdominal aortic aneurysm can be risky.

The ideal way to clean your teeth is no mystery; even small changes in your home dental care can lead longer-lasting teeth. Typically, by age of fifty, you will have crowns, bridges, partial prostheses, and sometimes, even a full prosthesis

Penile Injection Therapy

Have you heard any medical lingo you've thought is strange? Funny healthcare speaker Dr. Brad Nieder discusses funny medical terminology he's learned in his medical career. He brings his medical comedy to a healthcare conference, describing how he didn't know what "stat" meant.
He goes on about how he thought up many funny terms he could say in return to the doctor who introduced him to the word. His healthcare comedy makes the crowd burst with laughter.
Dr. Brad knows how to adapt his hilarious real-life stories into customized presentations for any in-person or virtual event. Watch more of his videos as a medical comedian and all-around funny guy by browsing his videos.

UPDATE 1/30/15: Watch the updated version of this animation: https://www.youtube.com/watch?v=LVP6JngpgEE
This 3D medical animation shows how adhesions in the abdomen may cause complications. These problems may include obstruction, twisting, and dislocating areas of the small intestine. Adhesions can be separated with laparoscopic instruments.
ANH00037
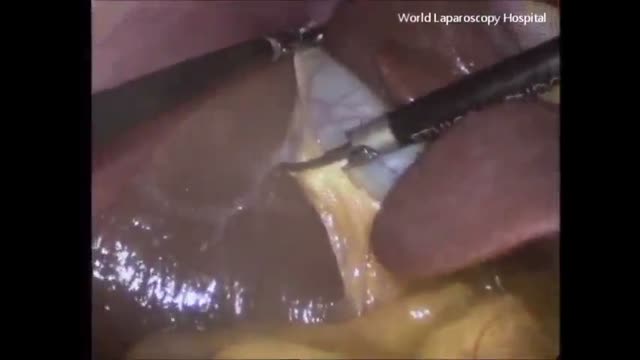
A high definition HD video of Laparoscopic Cholecystectomy surgery
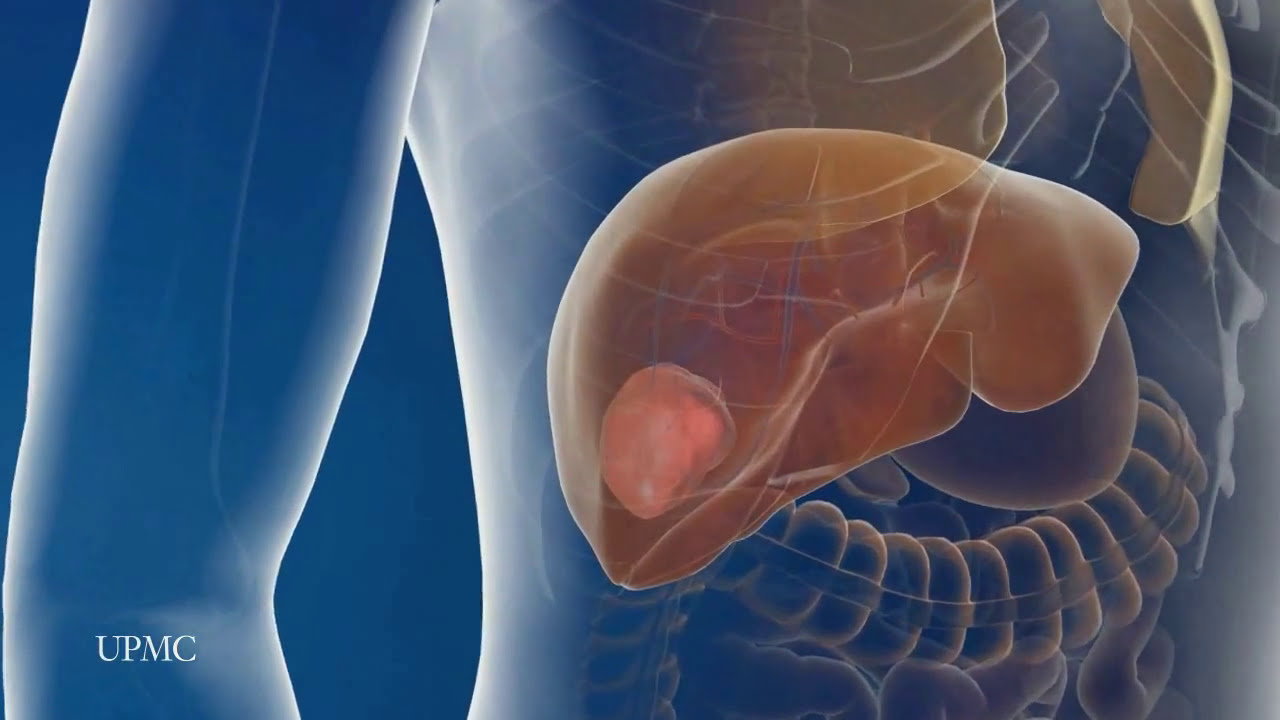
UPMC liver surgeons are among the most experienced in the world in performing minimally invasive liver surgery. Most patients benefit from less trauma and pain, minimal scarring, a shorter hospital stay, and faster recovery than from traditional surgery.
To learn more, please visit https://www.upmc.com/services/....liver-cancer/treatme

When being overweight becomes more than just an inconvenience.

In breech position, the baby's bottom is down. There are a few types of breech: Complete breech means the baby is bottom-first, with knees bent. Frank breech means the baby's legs are stretched up, with feet near the head. Footling breech means one leg is lowered over the mother's cervix. You are more likely to have a breech baby if you: Go into early labor Have an abnormally shaped uterus, fibroids, or too much amniotic fluid Have more than one baby in your womb Have placenta previa (when the placenta is on the lower part of the uterine wall, blocking the cervix)
Panic attack from Injection:'(

HD Gynecomastia Surgery
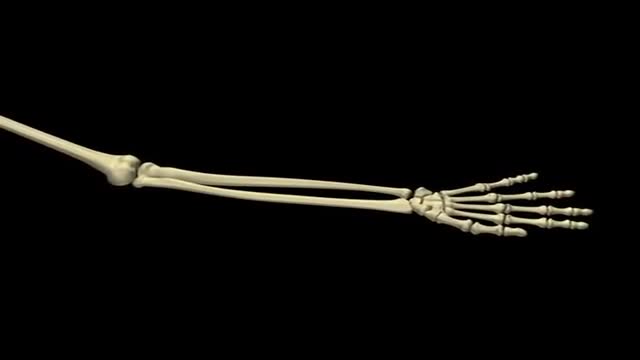
Carpal Tunnel Syndrom 3D Animation
Depression is a very serious mental illness that affects millions worldwide. Could a small brain implant cure it?
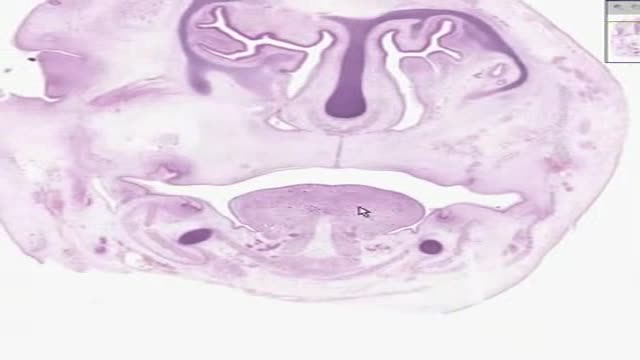
Histology of Tooth Development
